Protein markers for drug responses (IC50 screen)
Junyan Lu
2020-06-08
Last updated: 2020-06-09
Checks: 5 2
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- unnamed-chunk-12
- unnamed-chunk-17
- unnamed-chunk-22
- unnamed-chunk-7
To ensure reproducibility of the results, delete the cache directory analysisDrugResponses_IC50_cache and re-run the analysis. To have workflowr automatically delete the cache directory prior to building the file, set delete_cache = TRUE when running wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/analysisDrugResponses_IC50_cache/
Ignored: analysis/analysisDrugResponses_cache/
Ignored: analysis/complexAnalysis_IGHV_alternative_cache/
Ignored: analysis/complexAnalysis_IGHV_cache/
Ignored: analysis/complexAnalysis_trisomy12_alteredPQR_cache/
Ignored: analysis/complexAnalysis_trisomy12_alternative_cache/
Ignored: analysis/complexAnalysis_trisomy12_cache/
Ignored: analysis/correlateCLLPD_cache/
Ignored: analysis/predictOutcome_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/CNVanalysis_11q.Rmd
Untracked: analysis/CNVanalysis_trisomy12.Rmd
Untracked: analysis/CNVanalysis_trisomy19.Rmd
Untracked: analysis/analysisDrugResponses.Rmd
Untracked: analysis/analysisDrugResponses_IC50.Rmd
Untracked: analysis/analysisPCA.Rmd
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/complexAnalysis_IGHV.Rmd
Untracked: analysis/complexAnalysis_IGHV_alternative.Rmd
Untracked: analysis/complexAnalysis_overall.Rmd
Untracked: analysis/complexAnalysis_trisomy12.Rmd
Untracked: analysis/complexAnalysis_trisomy12_alternative.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/default.css
Untracked: analysis/del11q.pdf
Untracked: analysis/del11q_norm.pdf
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/plotExpressionCNV.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/style.css
Untracked: analysis/trisomy12.pdf
Untracked: analysis/trisomy12_AFcor.Rmd
Untracked: analysis/trisomy12_norm.pdf
Untracked: code/AlteredPQR.R
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/example_proteomics_data
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/proteins_in_complexes
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/LUMOS_processed.RData
Untracked: output/cnv_plots.zip
Untracked: output/cnv_plots/
Untracked: output/cnv_plots_norm.zip
Untracked: output/dxdCLL.RData
Untracked: output/exprCNV.RData
Untracked: output/lassoResults_CPS.RData
Untracked: output/lassoResults_IC50.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/plotCNV_allChr11_diff.pdf
Untracked: output/plotCNV_del11q_sum.pdf
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_LUMOS_20200430.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/splicingResults.RData
Untracked: output/timsTOF_processed.RData
Untracked: plotCNV_del11q_diff.pdf
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateCLLPD.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Correlations between protein abundance and drug response
Preprocess datasets
viabMat.auc <- ic50 %>% filter(! Drug %in% c("DMSO","PBS"), patientID %in% colnames(protCLL)) %>%
group_by(patientID, Drug) %>% summarise(viab = mean(normVal_auc)) %>%
spread(key = patientID, value = viab) %>%
data.frame(stringsAsFactors = FALSE) %>% column_to_rownames("Drug") %>%
as.matrix()
viabMat <- ic50 %>% filter(! Drug %in% c("DMSO","PBS"), patientID %in% colnames(protCLL)) %>%
group_by(patientID, Drug, concIndex) %>% summarise(viab = mean(normVal)) %>% ungroup() %>%
spread(key = patientID, value = viab) %>%
mutate(drugConc = paste0(Drug, "_",concIndex)) %>% select(-Drug, -concIndex) %>%
data.frame(stringsAsFactors = FALSE) %>% column_to_rownames("drugConc") %>%
as.matrix()Proteomics data
proMat <- assays(protCLL)[["count"]]
proMat <- proMat[,colnames(viabMat)]Remove proteins without much variance (to lower multi-testing burden)
sds <- genefilter::rowSds(proMat,na.rm=TRUE)
proMat <- proMat[sds > genefilter::shorth(sds),]Remove drugs without much variance (only for individual concentrations)
#individual concentrations
sds <- genefilter::rowSds(viabMat)
viabMat <- viabMat[sds > genefilter::shorth(sds),]How many samples have both proteomics data and ic50 screen data
ncol(proMat)[1] 32Association test without any blocking
For individual concentrations
Association test
resTab <- lapply(rownames(viabMat),function(drugName) {
viab <- viabMat[drugName, ]
designMat <- model.matrix(~1+viab)
fit <- lmFit(proMat, designMat)
fit2 <- eBayes(fit)
corRes <- topTable(fit2, number ="all", adjust.method = "BH", coef = "viab") %>% rownames_to_column("id") %>%
mutate(symbol = rowData(protCLL[id,])$hgnc_symbol, drugConc = drugName)
}) %>% bind_rows() %>% separate(drugConc, c("Drug","concIndex"),"_",remove = FALSE) %>%
mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>% arrange(P.Value)Number of associations (25% FDR)
Here I use an FDR cut-off of 25%, because if I use 10%, there will be only 2 associations.
Select significant associations
resTab.sig <- filter(resTab, adj.P.Val < 0.25) %>%
select(Drug, symbol, id,logFC, P.Value, adj.P.Val, concIndex)plotTab <- resTab.sig %>% group_by(Drug, concIndex) %>%
summarise(n = length(id)) %>% ungroup()
ordTab <- group_by(plotTab, Drug) %>% summarise(total = sum(n)) %>%
arrange(desc(total))
plotTab <- mutate(plotTab, Drug = factor(Drug, levels = ordTab$Drug)) %>%
filter(n>0)
ggplot(plotTab, aes(x=Drug,y=n,fill = concIndex)) + geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90,hjust=1,vjust=0.5)) +
ylab("Number of associations") + xlab("")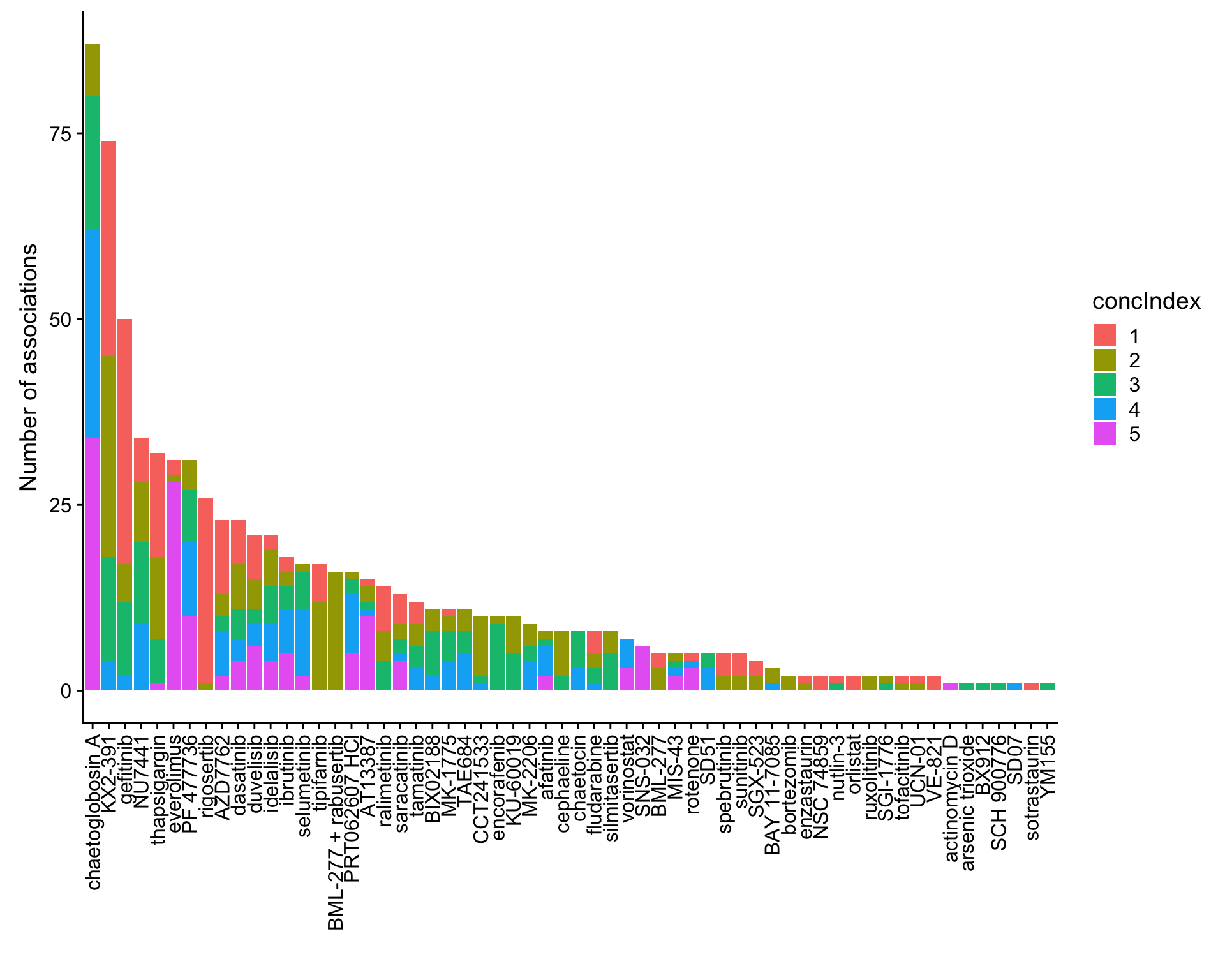
Table of significant associations
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()Plot top 9 drug-protein associations
plotList <- lapply(seq(9), function(i) {
drugConc <- paste0(resTab.sig$Drug[i],"_",resTab.sig$concIndex[i])
proteinName <- resTab.sig$symbol[i]
id <- resTab.sig$id[i]
plotTab <- tibble(patID = colnames(viabMat),
viab = viabMat[drugConc,],
expr = proMat[id,]) %>%
mutate(IGHV = protCLL[,patID]$IGHV.status,
trisomy12 = protCLL[,patID]$trisomy12)
ggplot(plotTab, aes(x=viab, y=expr)) + geom_point(aes(col = trisomy12, shape = IGHV)) +
scale_shape_manual(values = c(M = 19, U = 1)) + geom_smooth(method = "lm", se=FALSE) +
ggtitle(sprintf("%s ~ %s", drugConc, proteinName)) +
ylab("Protein expression") + xlab("Viability after treatment") +
theme(legend.position = "bottom")
})
plot_grid(plotlist = plotList, ncol =3)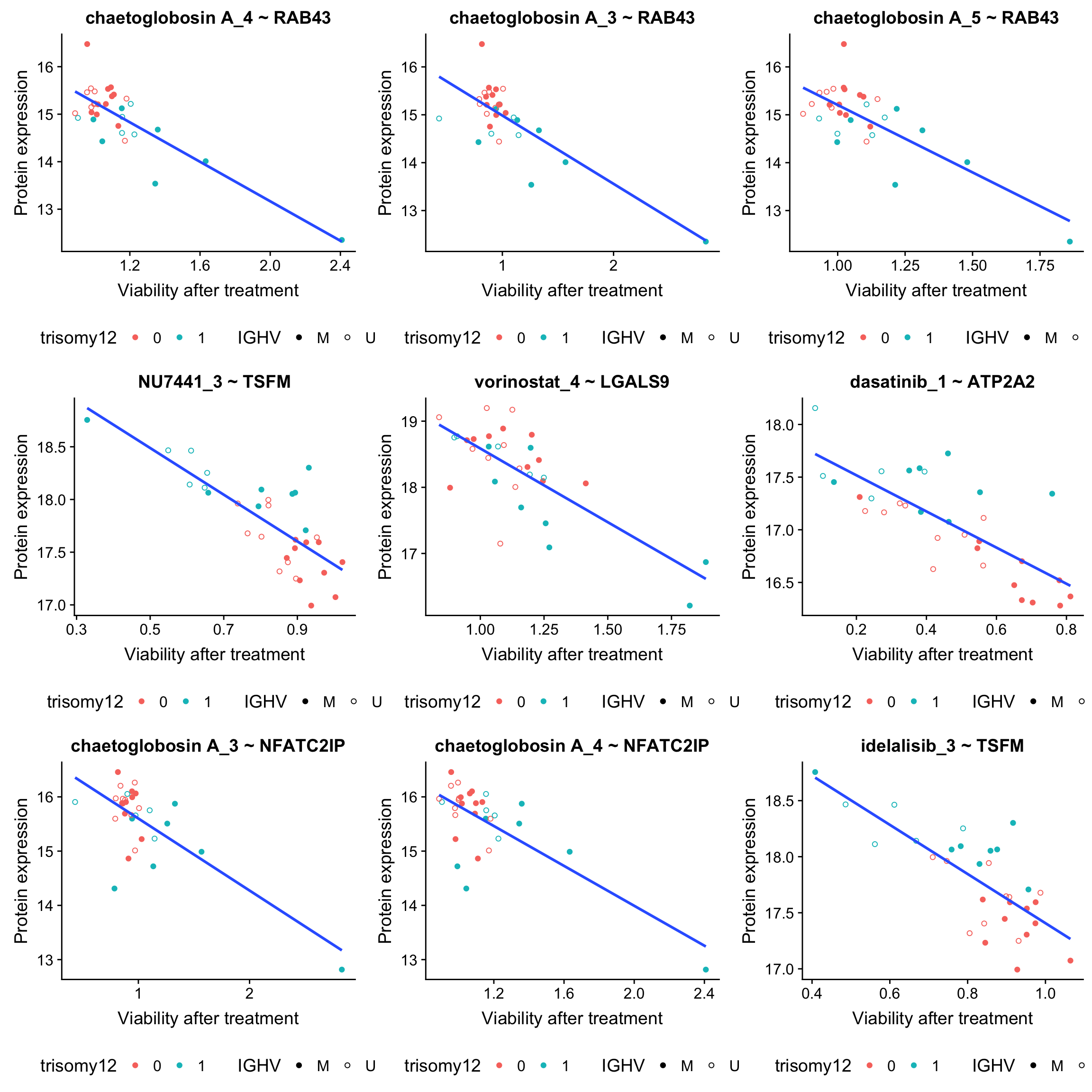 From those plots, it can be seen that those associations are potentially confounded by IGHV status and/or trisomy12.
From those plots, it can be seen that those associations are potentially confounded by IGHV status and/or trisomy12.
For averaged five concentrations (AUC)
Association test
resTab.auc <- lapply(rownames(viabMat.auc),function(drugName) {
viab <- viabMat.auc[drugName, ]
designMat <- model.matrix(~1+viab)
fit <- lmFit(proMat, designMat)
fit2 <- eBayes(fit)
corRes <- topTable(fit2, number ="all", adjust.method = "BH", coef = "viab") %>% rownames_to_column("id") %>%
mutate(symbol = rowData(protCLL[id,])$hgnc_symbol, Drug = drugName)
}) %>% bind_rows() %>% mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>% arrange(P.Value)Number of associations (25% FDR)
Select significant associations (25% FDR)
resTab.sig <- filter(resTab.auc, adj.P.Val < 0.25) %>%
select(Drug, symbol, id,logFC, P.Value, adj.P.Val)plotTab <- resTab.sig %>% group_by(Drug) %>%
summarise(n = length(id)) %>% ungroup()
ordTab <- group_by(plotTab, Drug) %>% summarise(total = sum(n)) %>%
arrange(desc(total))
plotTab <- mutate(plotTab, Drug = factor(Drug, levels = ordTab$Drug)) %>%
filter(n>0)
ggplot(plotTab, aes(x=Drug,y=n)) + geom_bar(stat = "identity", fill = "lightblue") +
theme(axis.text.x = element_text(angle = 90,hjust=1,vjust=0.5)) +
ylab("Number of associations") + xlab("")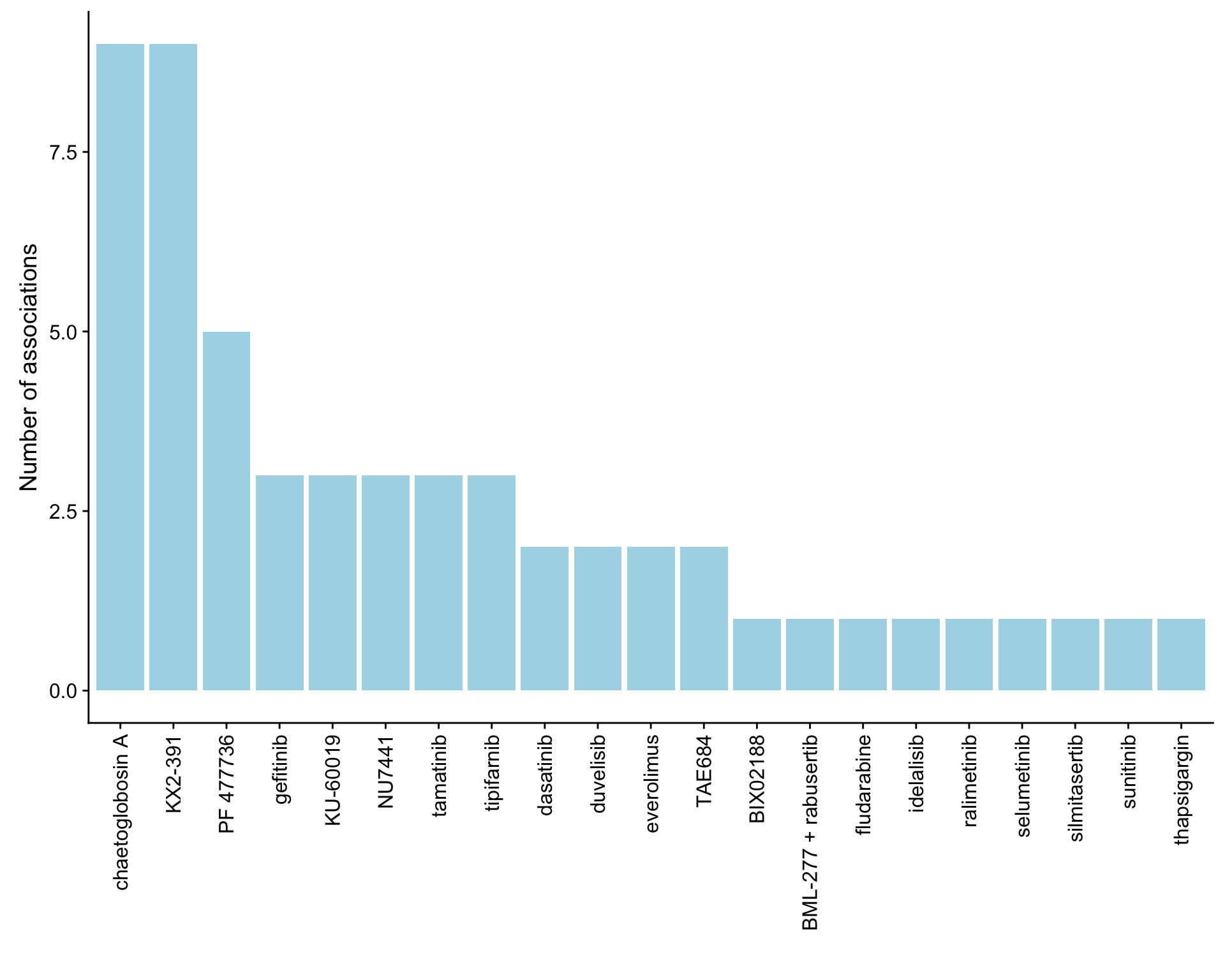
Table of significant associations
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()Plot top 9 drug-protein associations
plotList <- lapply(seq(9), function(i) {
drugConc <- resTab.sig$Drug[i]
proteinName <- resTab.sig$symbol[i]
id <- resTab.sig$id[i]
plotTab <- tibble(patID = colnames(viabMat.auc),
viab = viabMat.auc[drugConc,],
expr = proMat[id,]) %>%
mutate(IGHV = protCLL[,patID]$IGHV.status,
trisomy12 = protCLL[,patID]$trisomy12)
ggplot(plotTab, aes(x=viab, y=expr)) + geom_point(aes(col = trisomy12, shape = IGHV)) +
scale_shape_manual(values = c(M = 19, U = 1)) + geom_smooth(method = "lm", se=FALSE) +
ggtitle(sprintf("%s ~ %s", drugConc, proteinName)) +
ylab("Protein expression") + xlab("Viability after treatment") +
theme(legend.position = "bottom")
})
plot_grid(plotlist = plotList, ncol =3)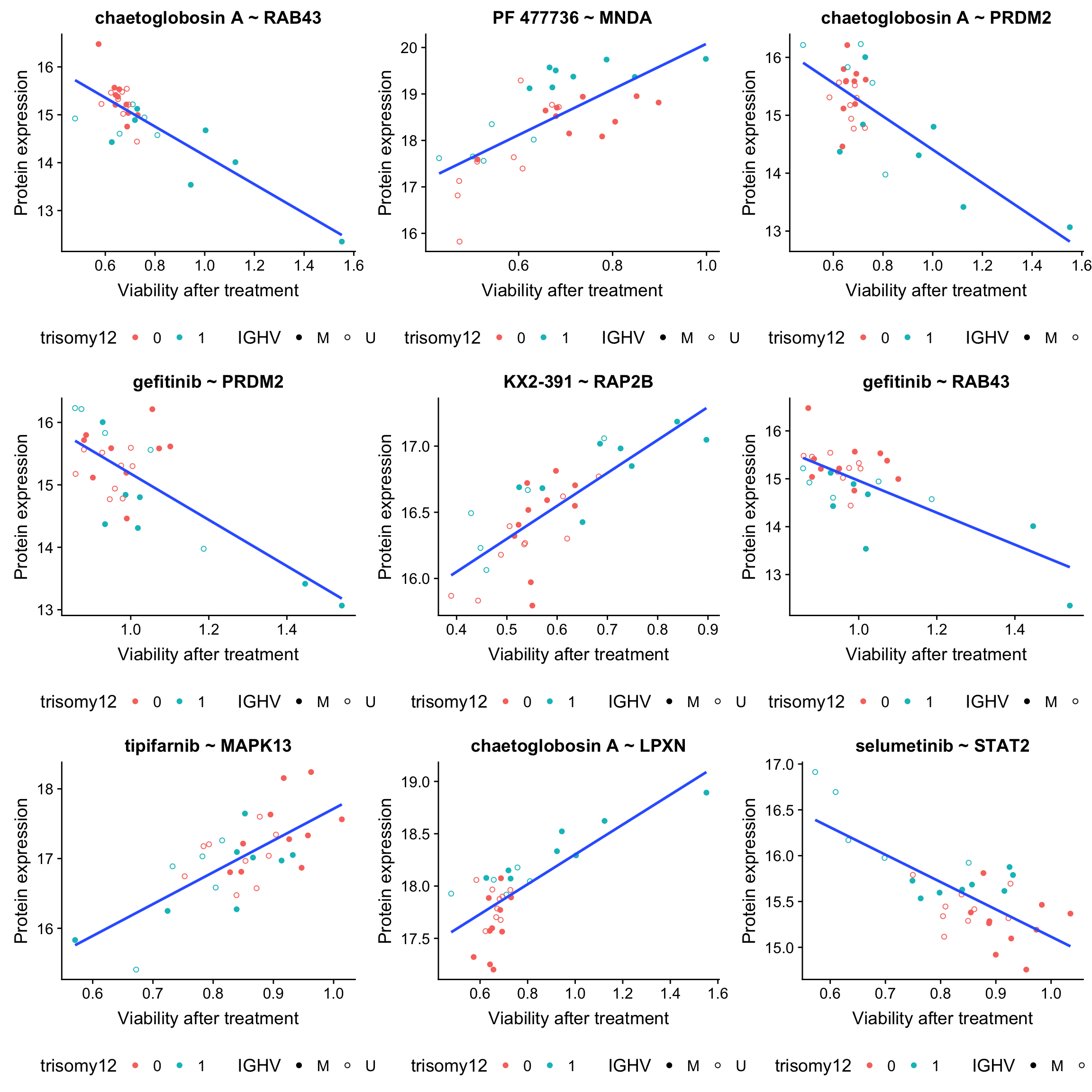
Association test with blocking for IGHV and trisomy12
For individual concentrations
Association test
testList <- filter(resTab, adj.P.Val < 0.25)
resTab.block <- lapply(seq(nrow(testList)),function(i) {
pair <- testList[i,]
expr <- proMat[pair$id,]
viab <- viabMat[pair$drugConc, ]
ighv <- protCLL[,colnames(viabMat)]$IGHV.status
tri12 <- protCLL[,colnames(viabMat)]$trisomy12
res <- anova(lm(viab~ighv+tri12+expr))
data.frame(id = pair$id, P.Value = res["expr",]$`Pr(>F)`, symbol = pair$symbol,
drugConc = pair$drugConc, Drug = pair$Drug, concIndex = pair$concIndex,
P.Value.IGHV = res["ighv",]$`Pr(>F)`,P.Value.trisomy12 = res["tri12",]$`Pr(>F)`,
P.Value.noBlock = pair$P.Value,
stringsAsFactors = FALSE)
}) %>% bind_rows() %>% mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>% arrange(P.Value)Number of associations (10% FDR)
Select significant associations
resTab.sig <- filter(resTab.block, adj.P.Val < 0.1) %>%
select(Drug, symbol, id, P.Value, adj.P.Val, P.Value.trisomy12, P.Value.IGHV, concIndex)plotTab <- resTab.sig %>% group_by(Drug, concIndex) %>%
summarise(n = length(id)) %>% ungroup()
ordTab <- group_by(plotTab, Drug) %>% summarise(total = sum(n)) %>%
arrange(desc(total))
plotTab <- mutate(plotTab, Drug = factor(Drug, levels = ordTab$Drug)) %>%
filter(n>0)
ggplot(plotTab, aes(x=Drug,y=n,fill = concIndex)) + geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90,hjust=1,vjust=0.5)) +
ylab("Number of associations") + xlab("")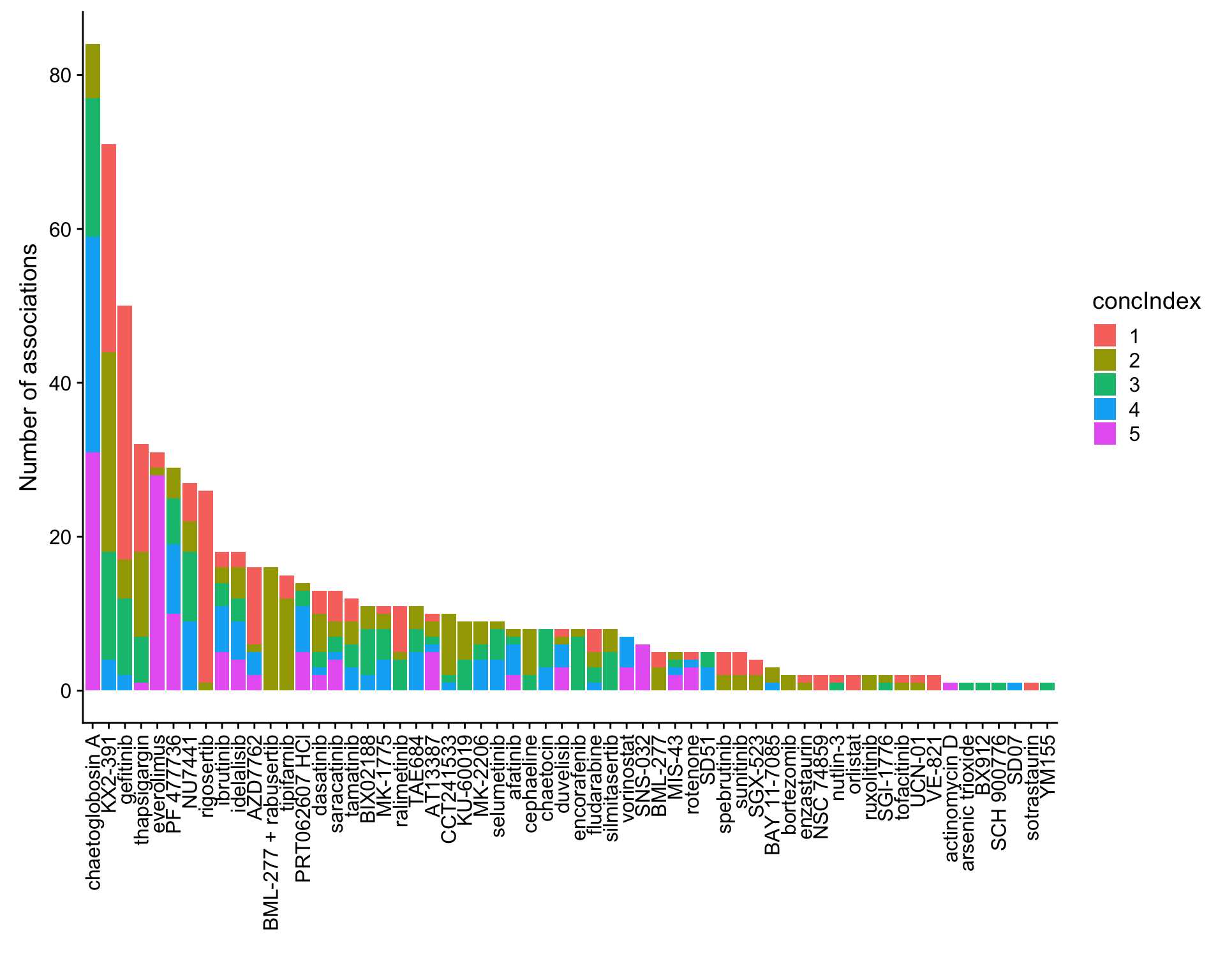
Table of significant associations
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()Plot top 9 drug-protein associations
plotList <- lapply(seq(9), function(i) {
drugConc <- paste0(resTab.sig$Drug[i],"_",resTab.sig$concIndex[i])
proteinName <- resTab.sig$symbol[i]
id <- resTab.sig$id[i]
plotTab <- tibble(patID = colnames(viabMat),
viab = viabMat[drugConc,],
expr = proMat[id,]) %>%
mutate(IGHV = protCLL[,patID]$IGHV.status,
trisomy12 = protCLL[,patID]$trisomy12)
ggplot(plotTab, aes(x=viab, y=expr,col = trisomy12, shape = IGHV, linetype = IGHV)) + geom_point() +
scale_shape_manual(values = c(M = 19, U = 1)) + geom_smooth(method = "lm", se=FALSE) +
ggtitle(sprintf("%s ~ %s", drugConc, proteinName)) +
ylab("Protein expression") + xlab("Viability after treatment") +
theme(legend.position = "bottom")
})
plot_grid(plotlist = plotList, ncol =3)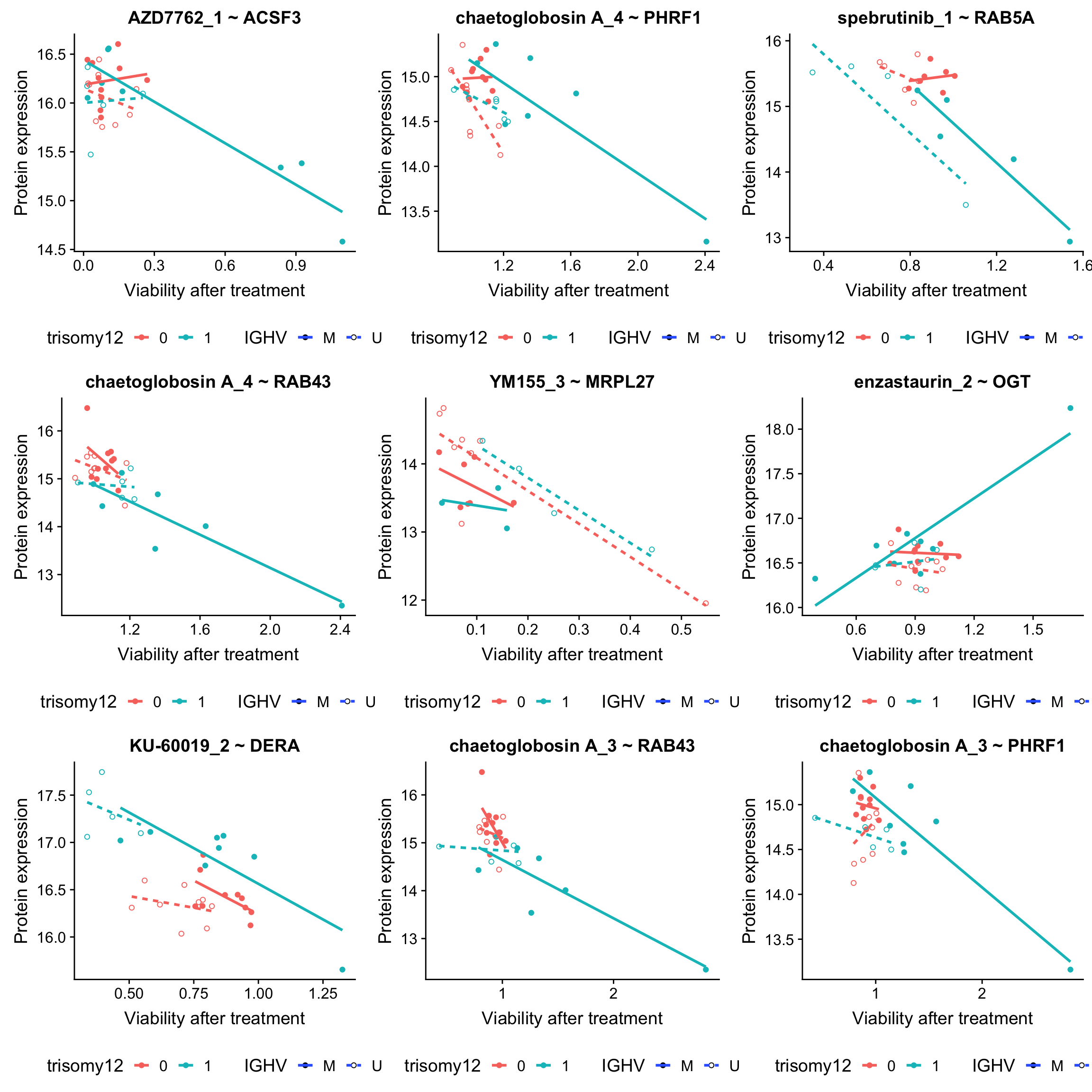
For averaged five concentrations (AUC)
Assocation test
testList <- filter(resTab.auc, adj.P.Val < 0.25)
resTab.auc.block <- lapply(seq(nrow(testList)),function(i) {
pair <- testList[i,]
expr <- proMat[pair$id,]
viab <- viabMat.auc[pair$Drug, ]
ighv <- protCLL[,colnames(viabMat.auc)]$IGHV.status
tri12 <- protCLL[,colnames(viabMat.auc)]$trisomy12
res <- anova(lm(viab~ighv+tri12+expr))
data.frame(id = pair$id, P.Value = res["expr",]$`Pr(>F)`, symbol = pair$symbol,
Drug = pair$Drug,
P.Value.IGHV = res["ighv",]$`Pr(>F)`,P.Value.trisomy12 = res["tri12",]$`Pr(>F)`,
P.Value.noBlock = pair$P.Value,
stringsAsFactors = FALSE)
}) %>% bind_rows() %>% mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>% arrange(P.Value)Number of associations (10% FDR)
Select significant associations
resTab.sig <- filter(resTab.auc.block, adj.P.Val < 0.1) %>%
select(Drug, symbol, id, P.Value, adj.P.Val, P.Value.trisomy12, P.Value.IGHV)plotTab <- resTab.sig %>% group_by(Drug) %>%
summarise(n = length(id)) %>% ungroup()
ordTab <- group_by(plotTab, Drug) %>% summarise(total = sum(n)) %>%
arrange(desc(total))
plotTab <- mutate(plotTab, Drug = factor(Drug, levels = ordTab$Drug)) %>%
filter(n>0)
ggplot(plotTab, aes(x=Drug,y=n)) + geom_bar(stat = "identity", fill = "lightblue") +
theme(axis.text.x = element_text(angle = 90,hjust=1,vjust=0.5)) +
ylab("Number of associations") + xlab("")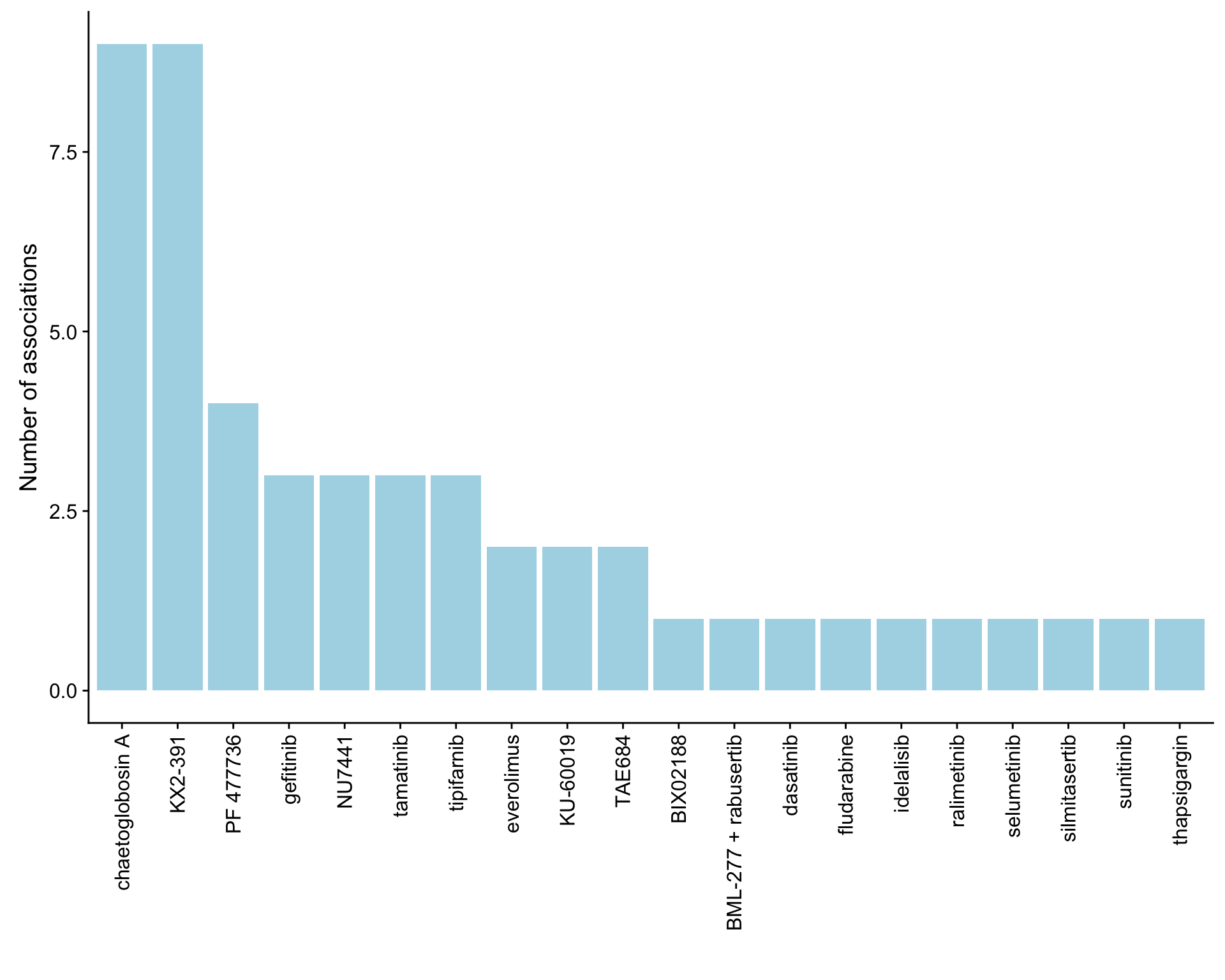
Table of significant associations (10% FDR)
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()Plot top 9 drug-protein associations
plotList <- lapply(seq(9), function(i) {
drugConc <- resTab.sig$Drug[i]
proteinName <- resTab.sig$symbol[i]
id <- resTab.sig$id[i]
plotTab <- tibble(patID = colnames(viabMat.auc),
viab = viabMat.auc[drugConc,],
expr = proMat[id,]) %>%
mutate(IGHV = protCLL[,patID]$IGHV.status,
trisomy12 = protCLL[,patID]$trisomy12)
ggplot(plotTab, aes(x=viab, y=expr,col = trisomy12, shape = IGHV, linetype = IGHV)) + geom_point(aes()) +
scale_shape_manual(values = c(M = 19, U = 1)) + geom_smooth(method = "lm", se=FALSE) +
ggtitle(sprintf("%s ~ %s", drugConc, proteinName)) +
ylab("Protein expression") + xlab("Viability after treatment") +
theme(legend.position = "bottom")
})
plot_grid(plotlist = plotList, ncol =3)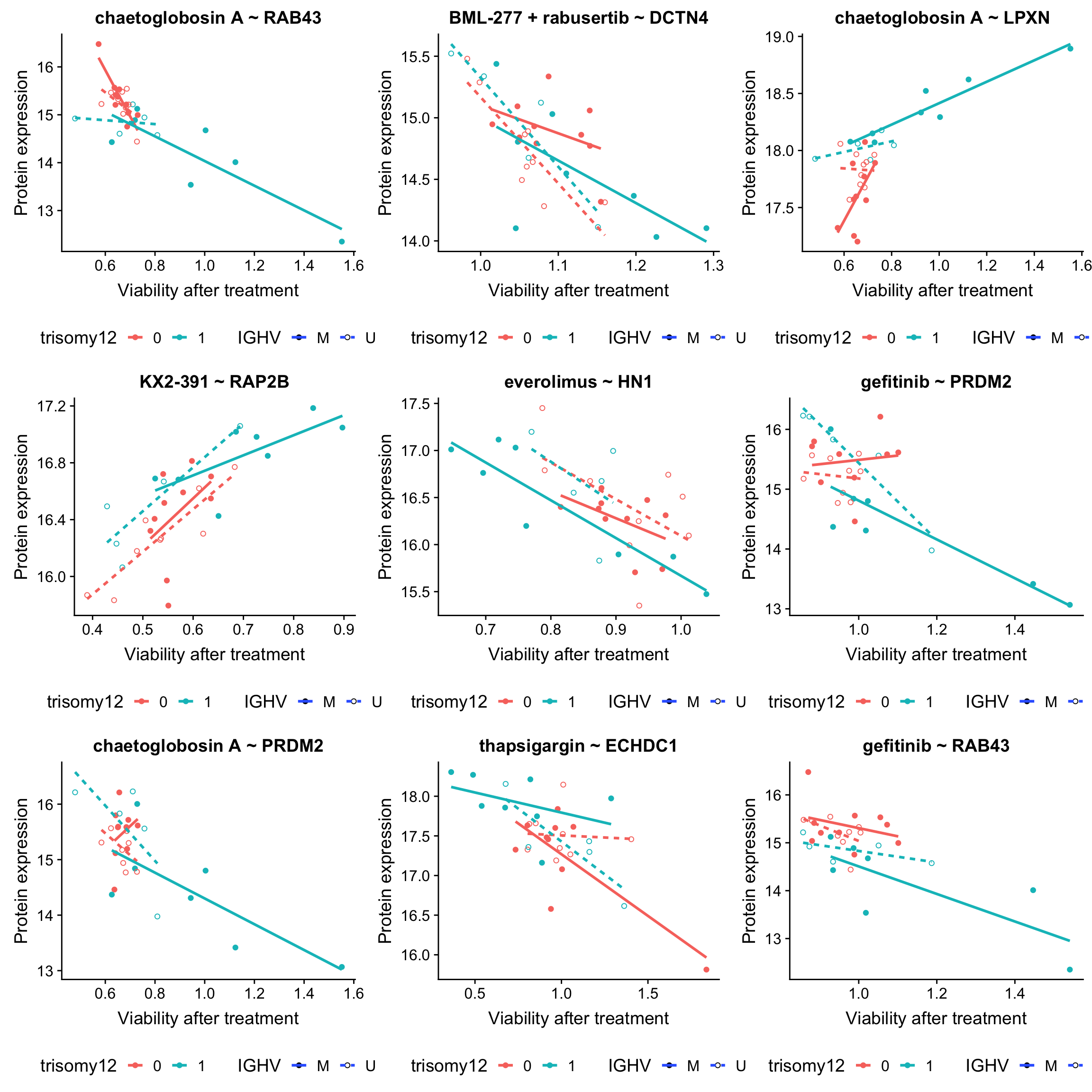
Compare the ability to explain drug response among genomic, RNA and protein data
Prepare data
Proteomics data
proMat <- assays(protCLL)[["QRILC"]]
proMat <- proMat[,colnames(viabMat.auc)]
sds <- genefilter::rowSds(proMat,na.rm=TRUE)
proMat <- proMat[sds > genefilter::shorth(sds),]removeCorrelated <- function(x, cutoff = 0.8, distance = "cosine", cluster_method = "ward.D2") {
# calculate distiance matrix
if (distance == "binary") {
#maybe also usefull is the input is a sparse matrix
distMat <- dist(t(x), method = "binary")
} else if (distance == "pearson") {
#otherwise, using pearson correlation
distMat <- as.dist(1-cor(x))
} else if (distance == "euclidean") {
distMat <- dist(t(x), method = "euclidean")
} else if (distance == "cosine") {
# cosine similarity maybe prefered for sparse matrix
cosineSimi <- function(x){
x%*%t(x)/(sqrt(rowSums(x^2) %*% t(rowSums(x^2))))
}
distMat <- as.dist(1-cosineSimi(t(x)))
} else if (distance == "canberra") {
distMat <- as.dist(as.matrix(dist(t(x), method = "canberra"))/nrow(x))
}
#hierarchical clustering
hc <- hclust(distMat, method = cluster_method)
clusters <- cutree(hc, h = 1-cutoff)
x.re <- x[,!duplicated(clusters)]
#record the removed features
mapList <- lapply(colnames(x.re), function(i) {
members <- names(clusters[clusters == clusters[i]])
members[members != i]
})
names(mapList) <- colnames(x.re)
return(list(reduced = x.re,
mapReduce = mapList))
}Remove highly correlated proteins
proReduced <- removeCorrelated(t(proMat), cutoff = 0.9, distance = "pearson")
proMat.re <- t(proReduced$reduced)Subset samples
overSample <- intersect(colnames(proMat.re), colnames(dds))
proMat.glm <- proMat.re[,overSample]
viabMat.glm <- viabMat.auc[,overSample]
dds.glm <- dds[,overSample]Prepare expression data
ddsSub.glm <- dds.glm[rowSums(counts(dds.glm)) > 100,]
ddsSub.glm <- varianceStabilizingTransformation(ddsSub.glm)
exprMat <- assay(ddsSub.glm)
sds <- genefilter::rowSds(exprMat)
exprMat <- exprMat[order(sds, decreasing=T)[1:5000],]
reduceRes <- removeCorrelated(t(exprMat), cutoff = 0.9, distance = "pearson")
exprMat.glm <- t(reduceRes$reduced)Prepare genomic data
geneMat <- patMeta[match(overSample, patMeta$Patient.ID),] %>%
select(Patient.ID, IGHV.status, del11p:U1) %>%
mutate_if(is.factor, as.character) %>% mutate(IGHV.status = ifelse(IGHV.status == "M", 1,0)) %>%
mutate_at(vars(-Patient.ID), as.numeric) %>% #assign a few unknown mutated cases to wildtype
data.frame() %>% column_to_rownames("Patient.ID")
geneMat <- geneMat[,apply(geneMat,2, function(x) sum(x %in% 1, na.rm = TRUE))>=3]
geneMat[is.na(geneMat)] <- 0Feature selection using LASSO
Prepare clean data: Integrate all available multi-omics datasets.
inclSet<-list(RNA=t(proMat.glm), drugs= t(viabMat.glm), Protein = t(proMat.glm), gen = geneMat)
cleanData <- generateData(inclSet, censor = 5)Perform lasso regression (3-fold repeated CV)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain)) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 20, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
save(lassoResults, file = "../output/lassoResults_IC50.RData", version = 2)Visualizing results
load("../output/lassoResults_IC50.RData")Variane explained (R2)
Averaged R2 for all drugs
#load save data
outList.train <- plotVar(lassoResults,cv=FALSE)
outList.cv <- plotVar(lassoResults, cv=TRUE)
sumTab.cv <- outList.cv$summary %>% mutate(group = "CV")
sumTab.train <- outList.train$summary %>% mutate(group = "train")
sumTab <- bind_rows(sumTab.train, sumTab.cv)
std <- function(x) sd(x)/sqrt(length(x))
plotTab <- sumTab %>%
group_by(set,group) %>% summarise(R2 = mean(meanR2), sem = std(meanR2))
ggplot(plotTab, aes(x=set, y = R2, fill = group)) + geom_bar(stat="identity", width = 0.5, position = "dodge") +
geom_errorbar(aes(ymin = R2 -sem, ymax=R2+sem), width= 0.5, position = "dodge") +
theme(legend.position = "none") + theme_bw()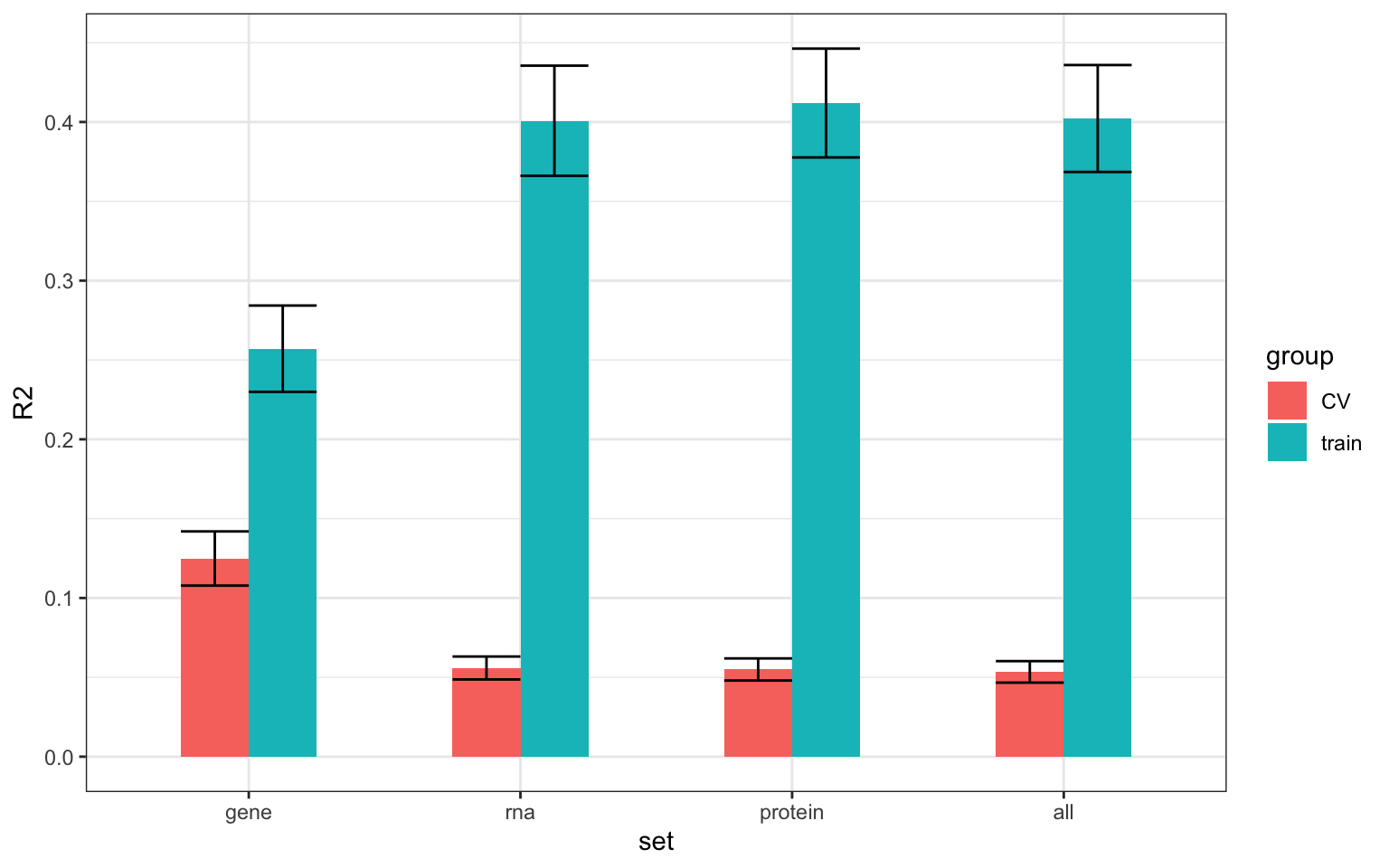
For individual drugs
plotTab <- filter(sumTab, set != "")
#rank by variance explained by proteomics data
drugRank <- plotTab %>% filter(set == "protein", group == "CV") %>% arrange(desc(meanR2)) %>% pull(drug)
plotList <- lapply(drugRank, function(name){
eachTab <- filter(plotTab, drug == name)
ggplot(eachTab,(aes(x=set, y = meanR2, fill = group))) +
geom_bar(stat ="identity",position = "dodge2", width=0.5) +
ggtitle(name) + coord_cartesian(ylim = c(0,1)) +
geom_errorbar(aes(ymax = meanR2 + sdR2, ymin = meanR2-sdR2), position = "dodge2", width=0.5) +
theme_bw() + theme(legend.position = "none")
})
plot_grid(plotlist = plotList, ncol =3)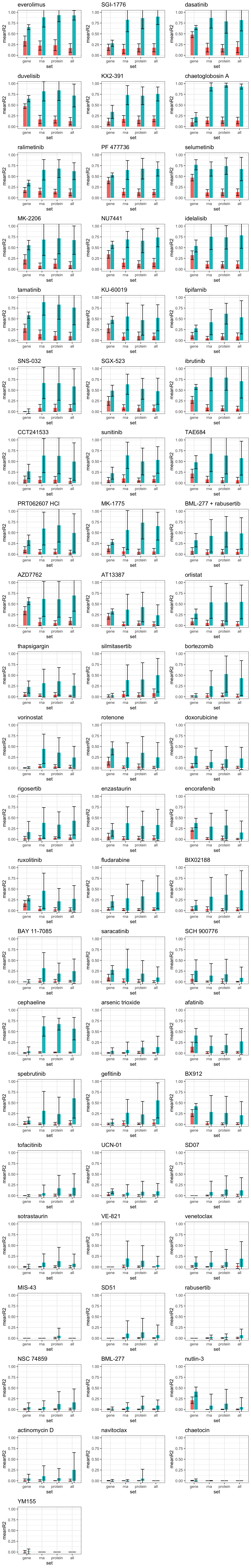
Proteins selected for each drug
Here I only use the top 10 drugs that best explained by proteomics
drugList <- drugRank[1:10]
plotList <- lapply(drugList,function(n) {
plotMat <- lassoResults[[n]]$protein$coefMat
plotMat <- plotMat[rowMeans(plotMat)!=0,]
plotTab <- plotMat %>% data.frame() %>% rownames_to_column("id") %>%
gather(key = "rep",value = "coef",-id) %>%
group_by(id) %>% summarise(meanCoef = mean(coef),semCoef=std(coef),freq=mean(sign(abs(coef)))) %>%
mutate(id=str_remove(id,"con.protein")) %>%
mutate(symbol = rowData(protCLL[id,])$hgnc_symbol) %>%
arrange(desc(meanCoef)) %>% mutate(symbol = factor(symbol, levels = symbol))
ggplot(plotTab, aes(x=symbol,y=meanCoef,fill = freq)) +
geom_bar(stat = "identity") + geom_errorbar(aes(ymax=meanCoef+semCoef, ymin = meanCoef-semCoef)) +
scale_fill_gradient(high="darkred", low="darkblue",name ="frequency", limits=c(0,1)) +
theme(legend.position = "right", axis.text.x = element_text(angle = 90, hjust=1, vjust = 0.5)) +
ggtitle(n) +
ylab("Coefficient") + xlab("")
})
plotList[[1]]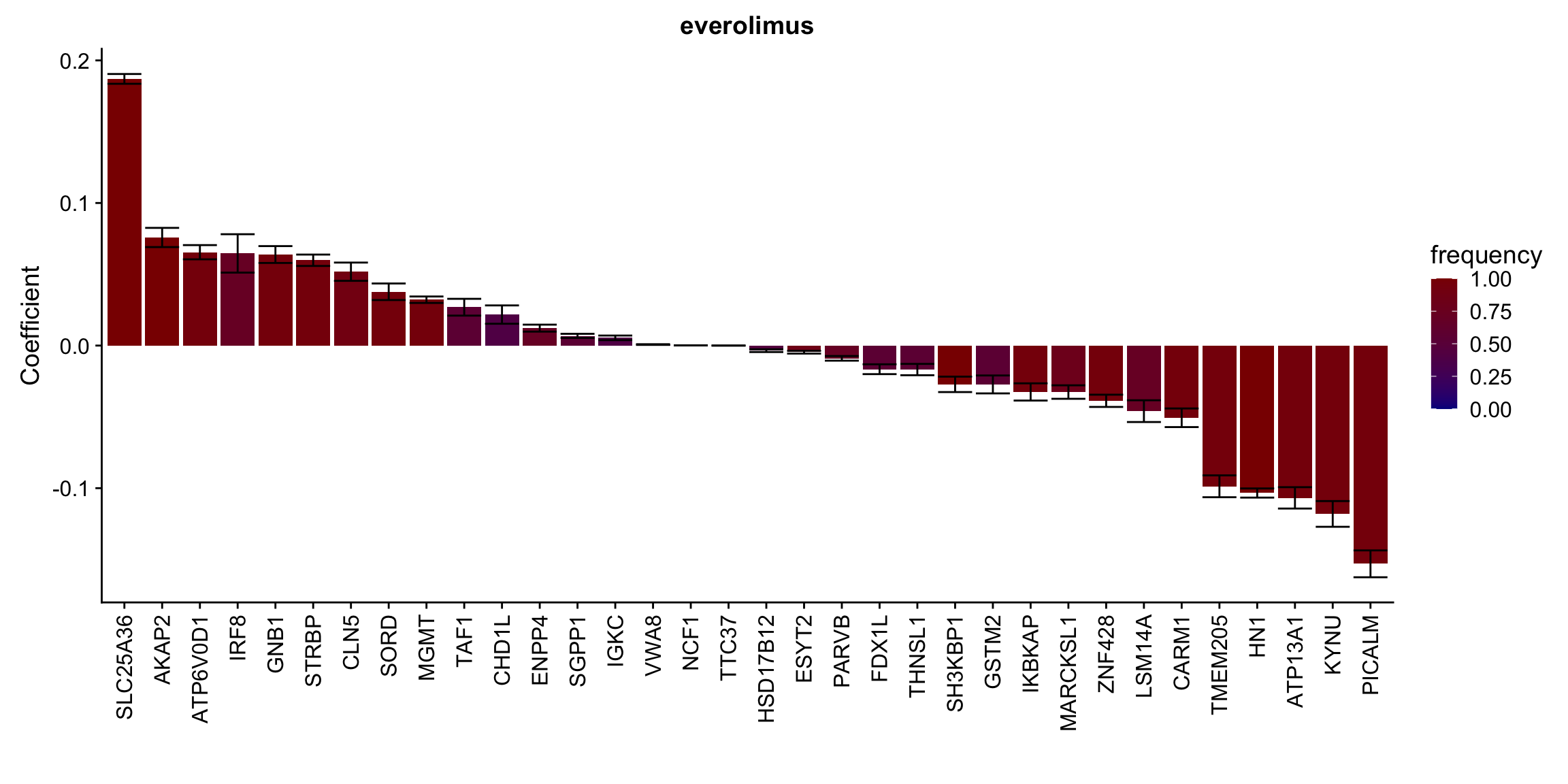
[[2]]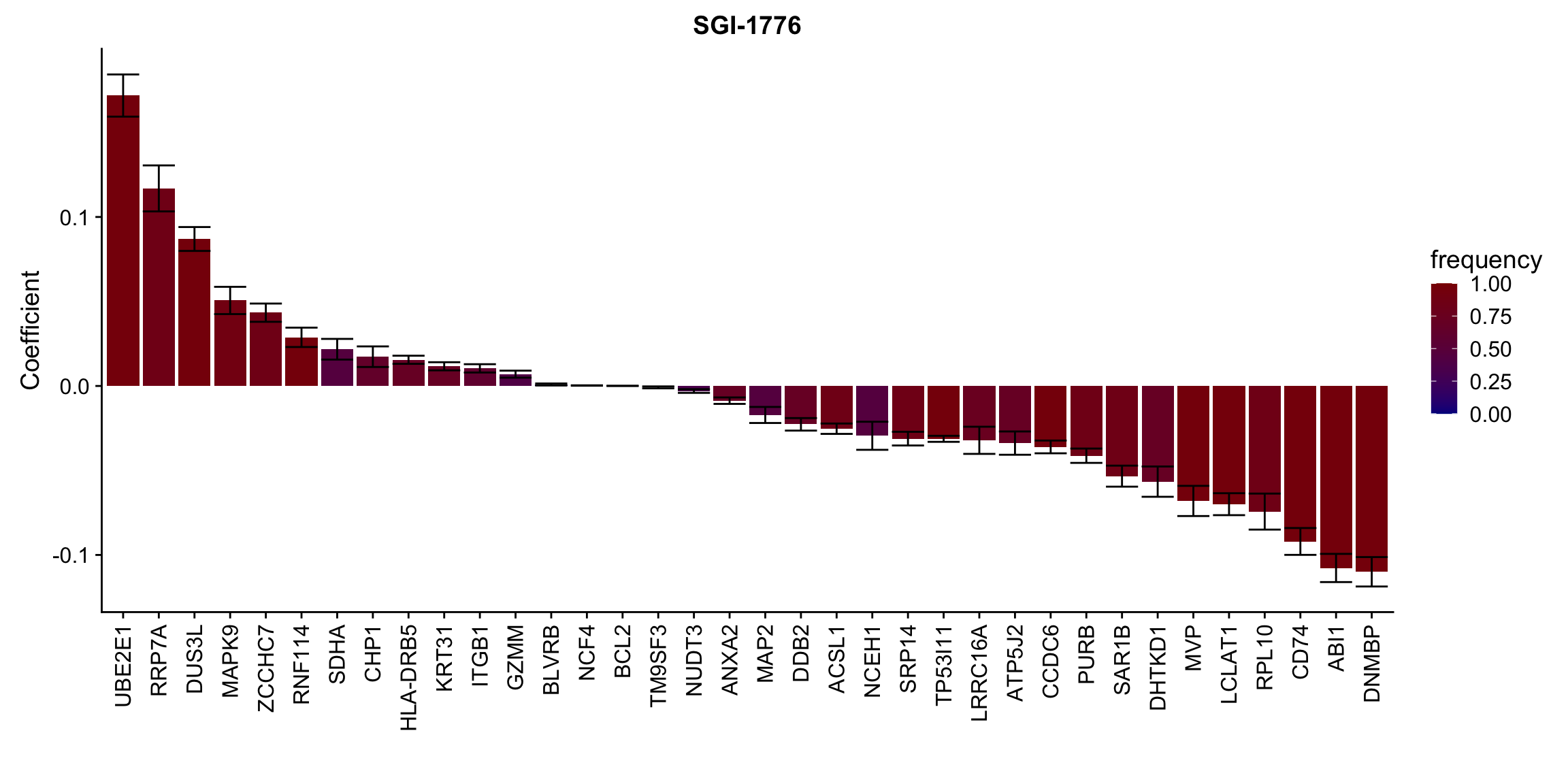
[[3]]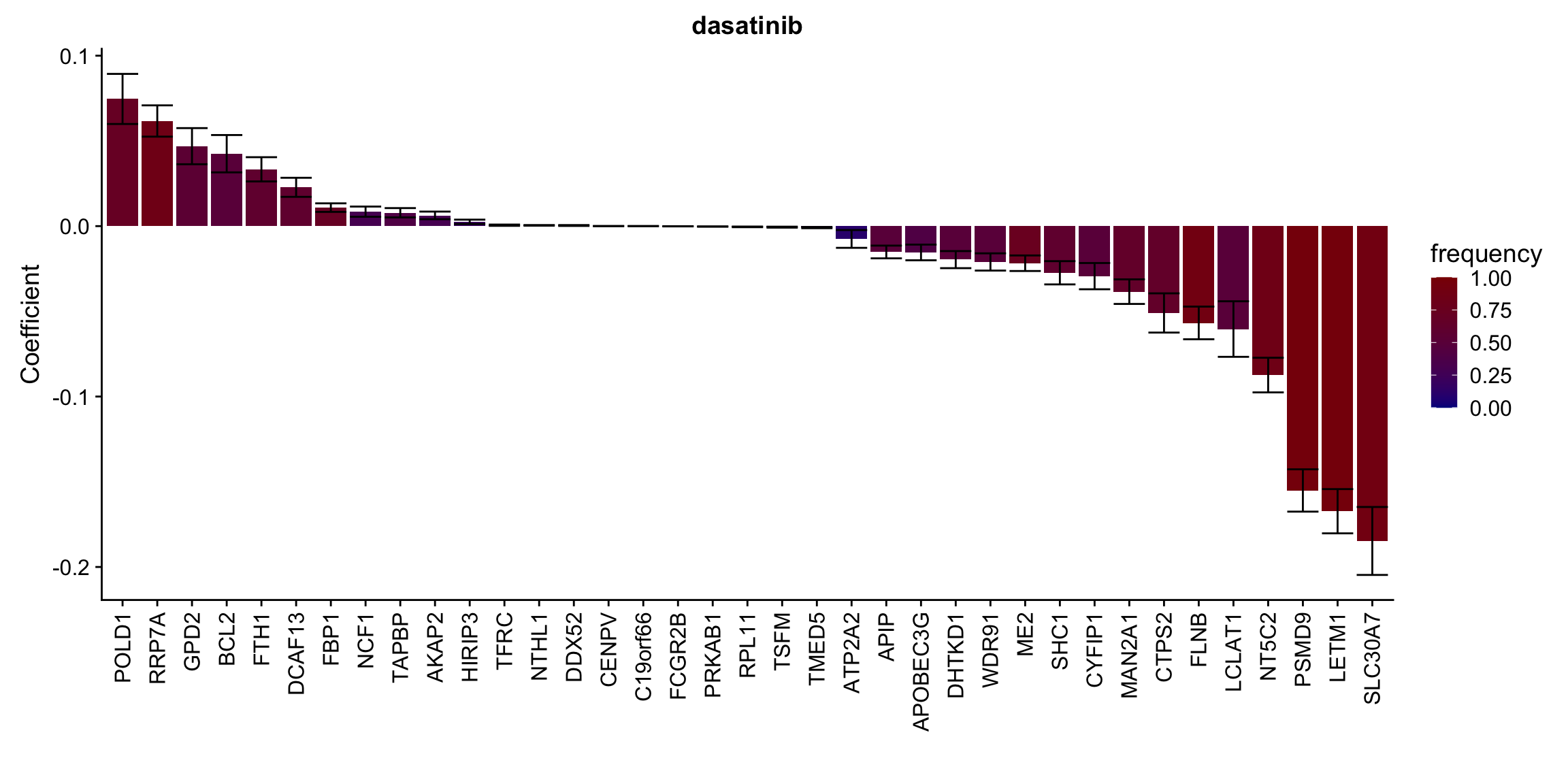
[[4]]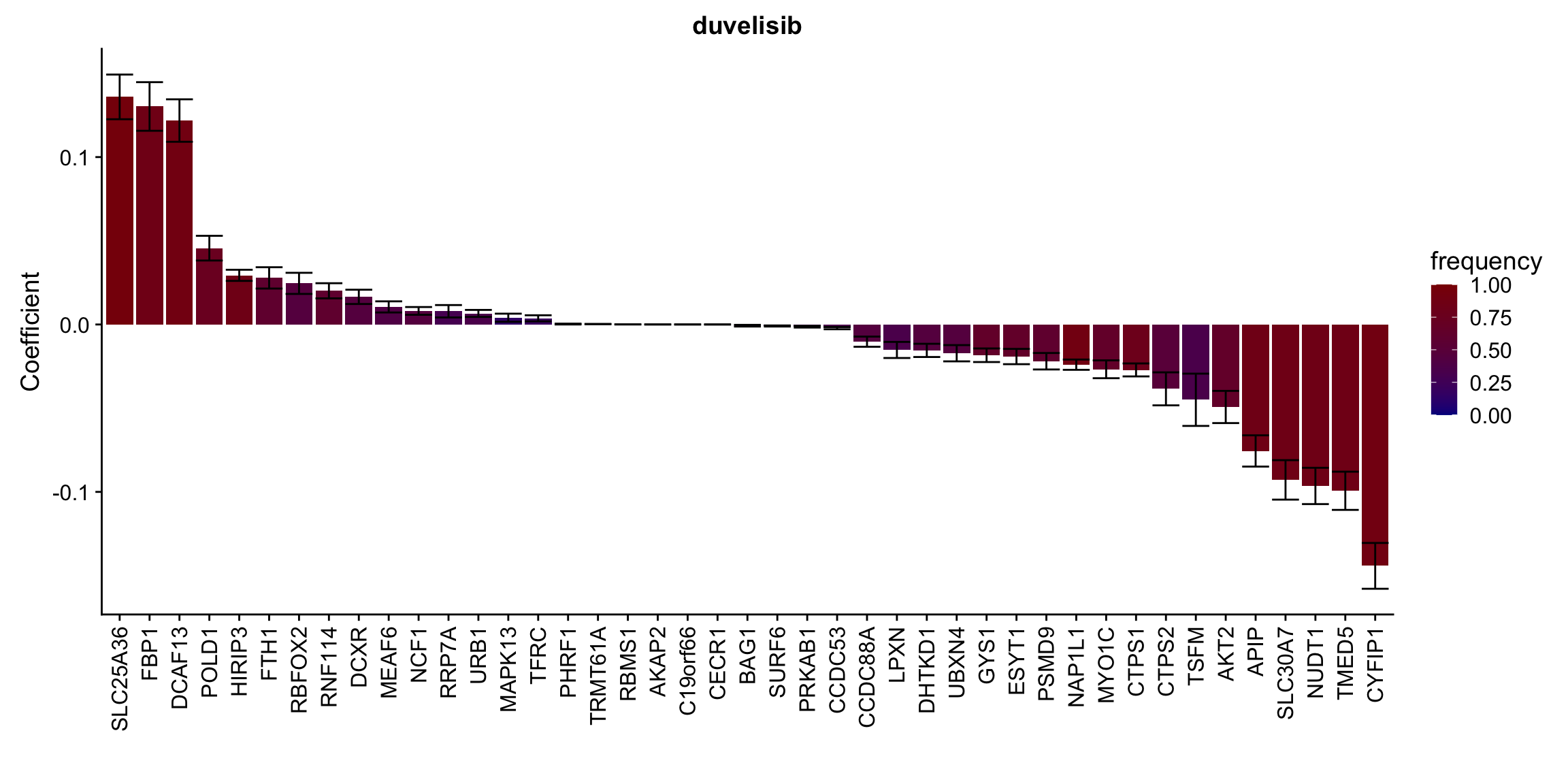
[[5]]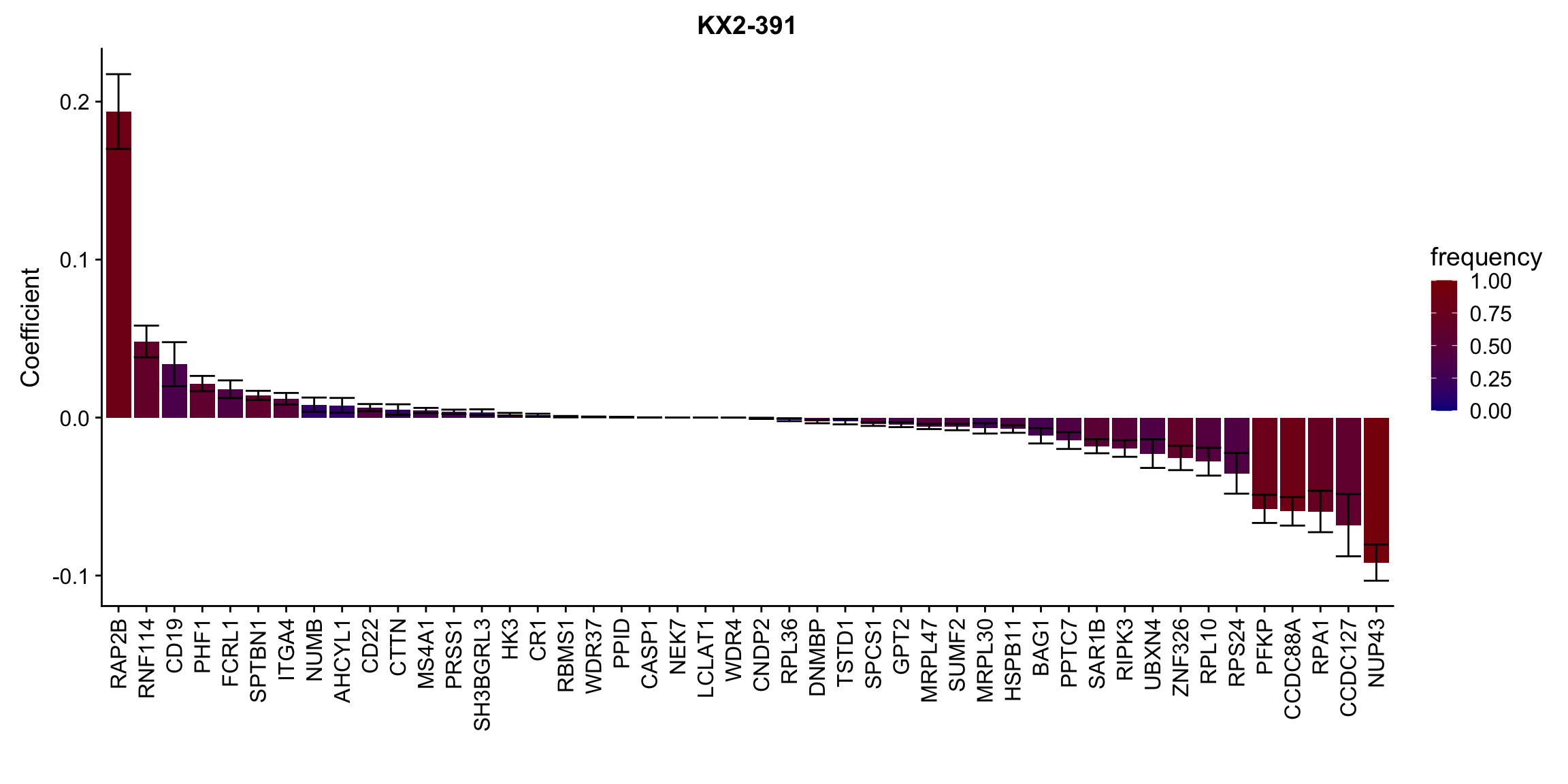
[[6]]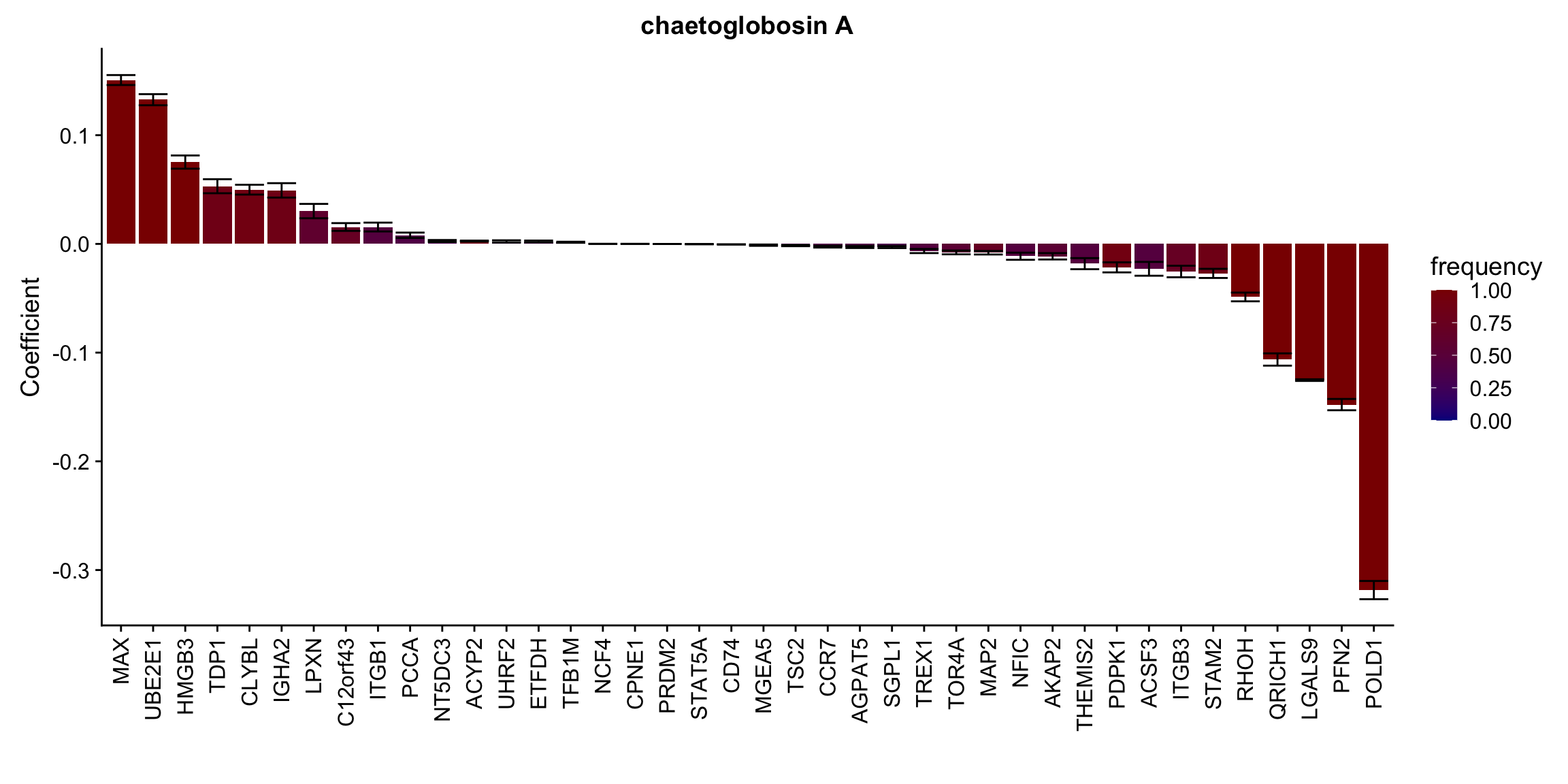
[[7]]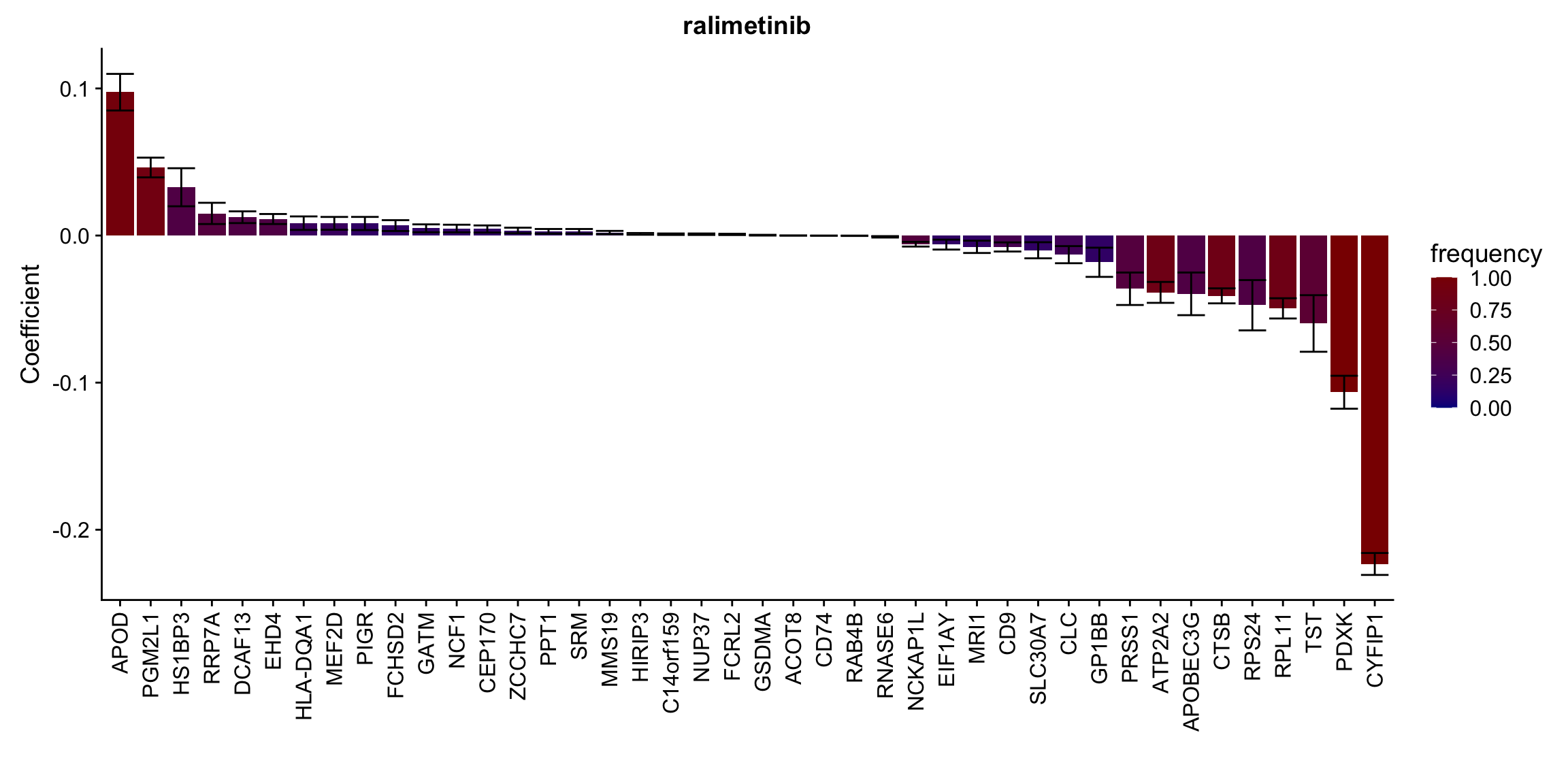
[[8]]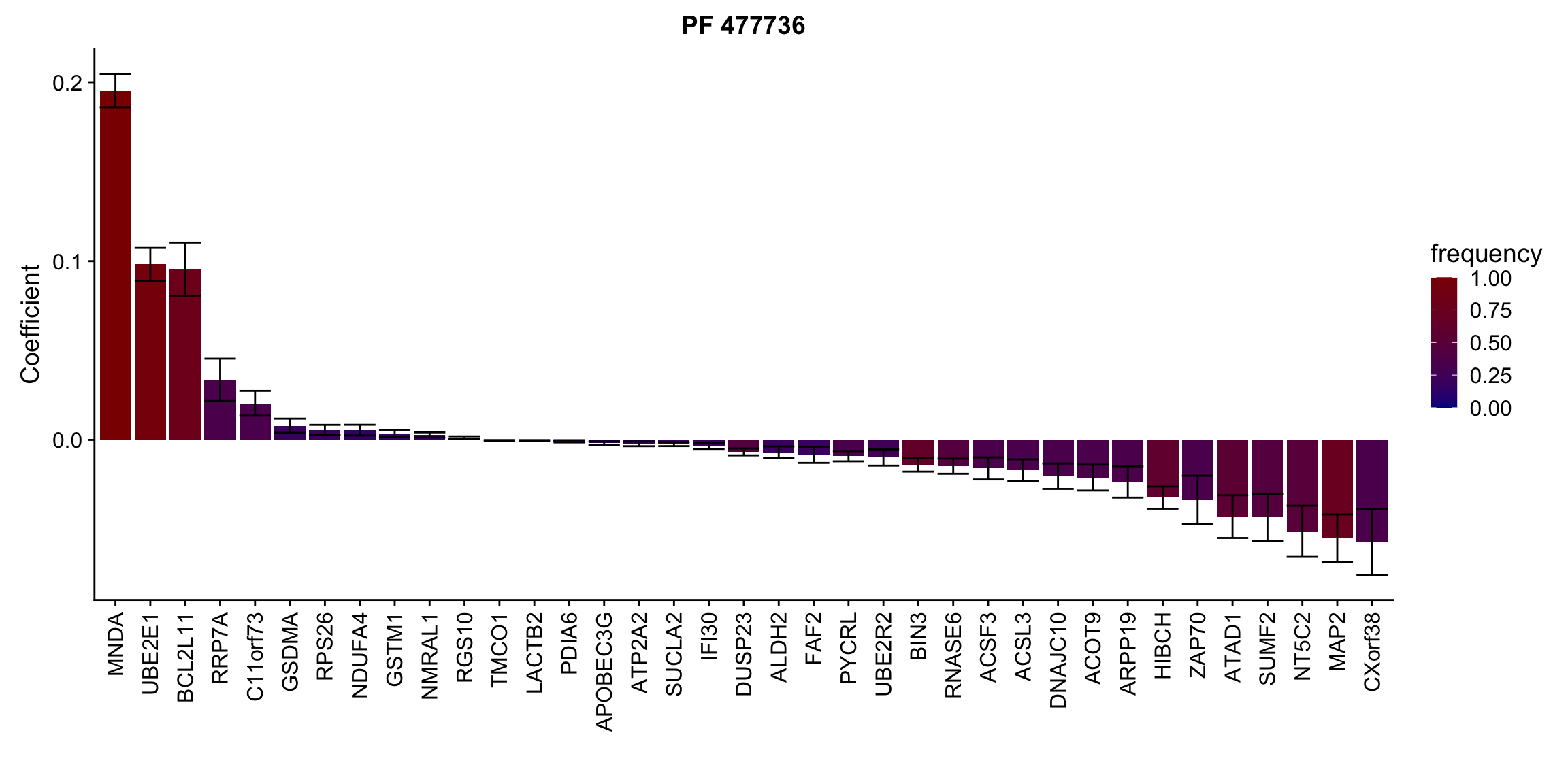
[[9]]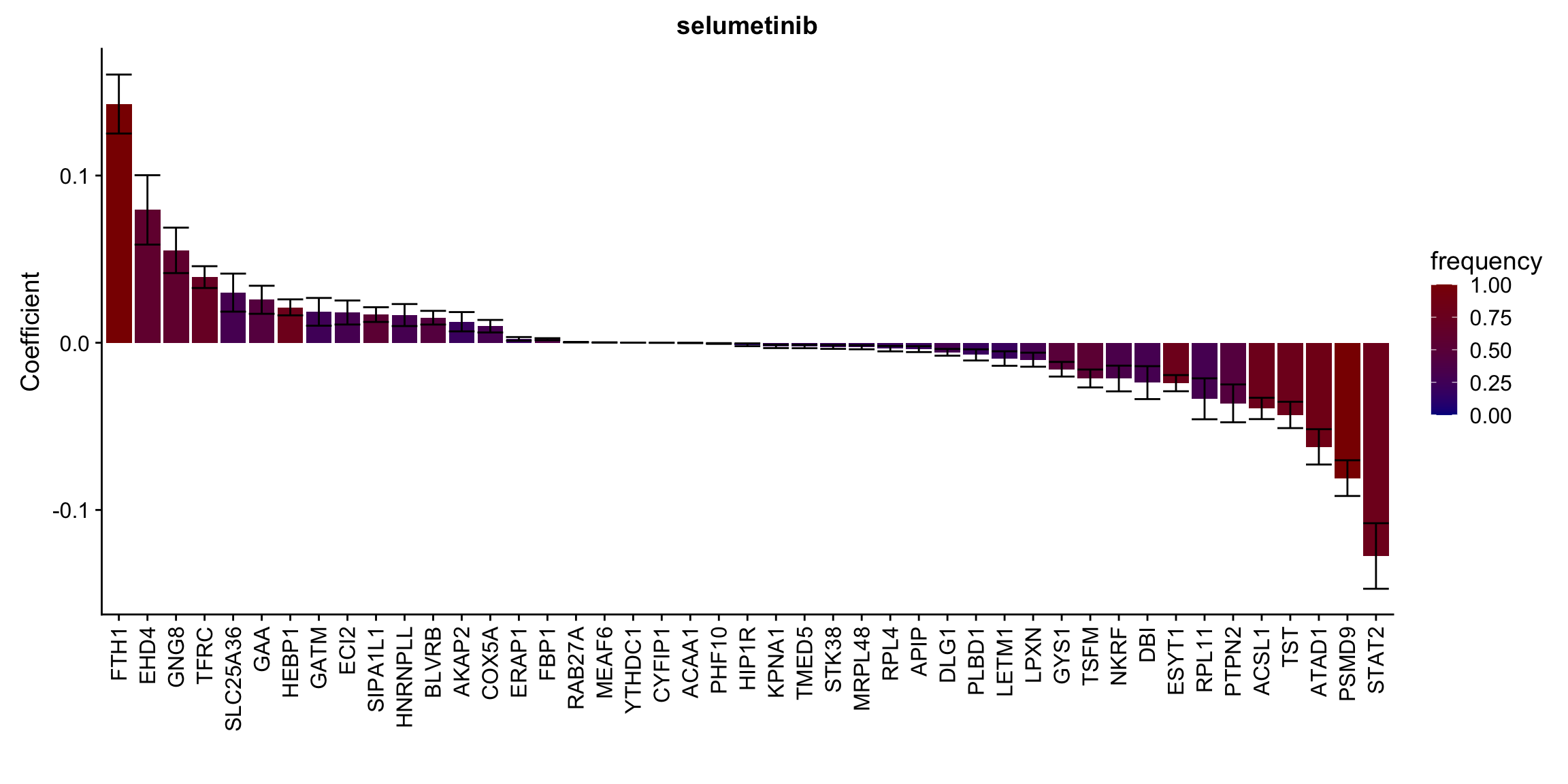
[[10]]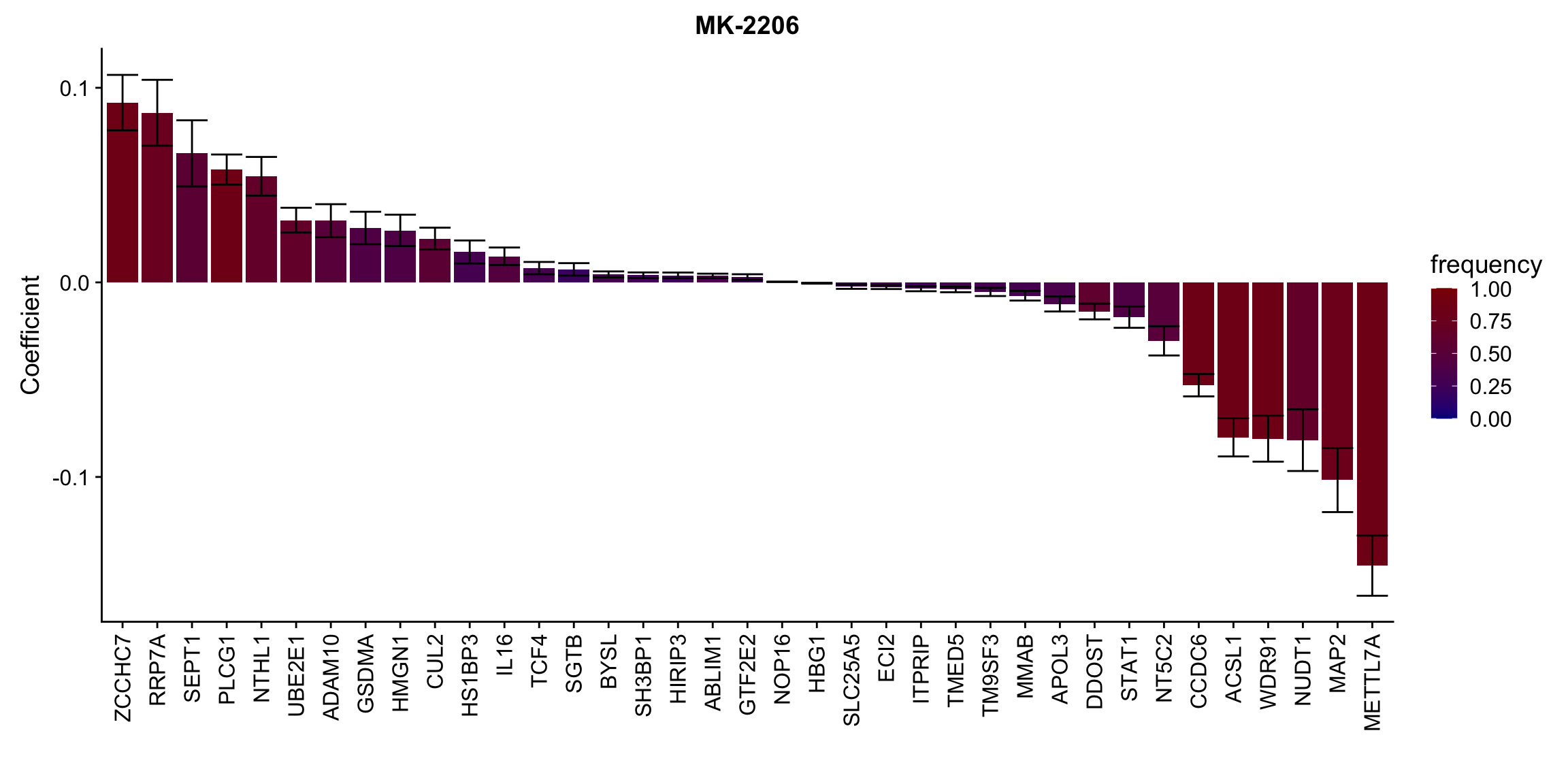
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DESeq2_1.24.0 forcats_0.4.0
[3] stringr_1.4.0 dplyr_0.8.5
[5] purrr_0.3.3 readr_1.3.1
[7] tidyr_1.0.0 tibble_3.0.0
[9] tidyverse_1.3.0 SummarizedExperiment_1.14.0
[11] DelayedArray_0.10.0 BiocParallel_1.18.0
[13] matrixStats_0.54.0 Biobase_2.44.0
[15] GenomicRanges_1.36.0 GenomeInfoDb_1.20.0
[17] IRanges_2.18.1 S4Vectors_0.22.0
[19] BiocGenerics_0.30.0 glmnet_2.0-18
[21] foreach_1.4.4 Matrix_1.2-17
[23] jyluMisc_0.1.5 pheatmap_1.0.12
[25] cowplot_0.9.4 ggplot2_3.3.0
[27] limma_3.40.2
loaded via a namespace (and not attached):
[1] shinydashboard_0.7.1 tidyselect_1.0.0 RSQLite_2.1.1
[4] AnnotationDbi_1.46.0 htmlwidgets_1.3 grid_3.6.0
[7] maxstat_0.7-25 munsell_0.5.0 codetools_0.2-16
[10] DT_0.7 withr_2.1.2 colorspace_1.4-1
[13] knitr_1.23 rstudioapi_0.10 ggsignif_0.5.0
[16] labeling_0.3 git2r_0.26.1 slam_0.1-45
[19] GenomeInfoDbData_1.2.1 KMsurv_0.1-5 bit64_0.9-7
[22] farver_2.0.3 rprojroot_1.3-2 vctrs_0.2.4
[25] generics_0.0.2 TH.data_1.0-10 xfun_0.8
[28] sets_1.0-18 R6_2.4.0 locfit_1.5-9.1
[31] bitops_1.0-6 fgsea_1.10.0 assertthat_0.2.1
[34] promises_1.0.1 scales_1.1.0 multcomp_1.4-10
[37] nnet_7.3-12 gtable_0.3.0 sandwich_2.5-1
[40] workflowr_1.6.0 rlang_0.4.5 genefilter_1.66.0
[43] cmprsk_2.2-8 splines_3.6.0 acepack_1.4.1
[46] broom_0.5.2 checkmate_2.0.0 yaml_2.2.0
[49] abind_1.4-5 modelr_0.1.5 crosstalk_1.0.0
[52] backports_1.1.4 httpuv_1.5.1 Hmisc_4.2-0
[55] tools_3.6.0 relations_0.6-8 ellipsis_0.2.0
[58] gplots_3.0.1.1 RColorBrewer_1.1-2 Rcpp_1.0.1
[61] base64enc_0.1-3 visNetwork_2.0.7 zlibbioc_1.30.0
[64] RCurl_1.95-4.12 ggpubr_0.2.1 rpart_4.1-15
[67] zoo_1.8-6 haven_2.2.0 cluster_2.1.0
[70] exactRankTests_0.8-30 fs_1.4.0 magrittr_1.5
[73] data.table_1.12.2 openxlsx_4.1.0.1 reprex_0.3.0
[76] survminer_0.4.4 mvtnorm_1.0-11 hms_0.5.2
[79] shinyjs_1.0 mime_0.7 evaluate_0.14
[82] xtable_1.8-4 XML_3.98-1.20 rio_0.5.16
[85] readxl_1.3.1 gridExtra_2.3 compiler_3.6.0
[88] KernSmooth_2.23-15 crayon_1.3.4 htmltools_0.4.0
[91] mgcv_1.8-28 later_0.8.0 Formula_1.2-3
[94] geneplotter_1.62.0 lubridate_1.7.4 DBI_1.0.0
[97] dbplyr_1.4.2 MASS_7.3-51.4 car_3.0-3
[100] cli_1.1.0 marray_1.62.0 gdata_2.18.0
[103] igraph_1.2.4.1 pkgconfig_2.0.2 km.ci_0.5-2
[106] foreign_0.8-71 piano_2.0.2 xml2_1.2.2
[109] annotate_1.62.0 XVector_0.24.0 drc_3.0-1
[112] rvest_0.3.5 digest_0.6.19 rmarkdown_1.13
[115] cellranger_1.1.0 fastmatch_1.1-0 survMisc_0.5.5
[118] htmlTable_1.13.1 curl_3.3 shiny_1.3.2
[121] gtools_3.8.1 lifecycle_0.2.0 nlme_3.1-140
[124] jsonlite_1.6 carData_3.0-2 pillar_1.4.3
[127] lattice_0.20-38 httr_1.4.1 plotrix_3.7-6
[130] survival_2.44-1.1 glue_1.3.2 zip_2.0.2
[133] iterators_1.0.10 bit_1.1-14 stringi_1.4.3
[136] blob_1.1.1 latticeExtra_0.6-28 caTools_1.17.1.2
[139] memoise_1.1.0