Analysis of differentially expressed proteins/RNAs related to trisomy19 and trisomy12
Junyan Lu
2020-03-13
Last updated: 2020-04-25
Checks: 6 1
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/correlateGenomic_PC12adjusted_cache/
Ignored: analysis/correlateGenomic_cache/
Ignored: analysis/correlateGenomic_noBlock_MCLL_cache/
Ignored: analysis/correlateGenomic_noBlock_UCLL_cache/
Ignored: analysis/correlateGenomic_noBlock_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/default.css
Untracked: analysis/patCNV.csv
Untracked: analysis/patCNV.xlsx
Untracked: analysis/patSNV.csv
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/style.css
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/LUMOS_processed.RData
Untracked: output/dxdCLL.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/splicingResults.RData
Untracked: output/timsTOF_processed.RData
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Proteomics data
Background of trisomy19 patients in the complete CLL cohort
tri19Tab <- filter(patMeta, diagnosis %in% "CLL") %>%
select(Patient.ID, IGHV.status, trisomy12, trisomy19) %>%
filter(trisomy19 %in% 1)
tri19Tab# A tibble: 7 x 4
Patient.ID IGHV.status trisomy12 trisomy19
<chr> <fct> <fct> <fct>
1 P0045 M 1 1
2 P0098 M 1 1
3 P0494 M 1 1
4 P0519 M 1 1
5 P0615 M 1 1
6 P0622 U 0 1
7 P0648 M 1 1 There are now 7 patients with trisomy19 in our cohort, most of them are M-CLL patients with trisomy12 One U-CLL patient, P0622, does not have trisomy12 but has trisomy19. This patients does not have FISH, WGS or WES data. The CNV status was inferred from methylation data
Differentially expressed proteins related to trisomy19 in M-CLL samples with trisomy12
Preprocessing
protCLL$trisomy19 <- patMeta[match(colnames(protCLL), patMeta$Patient.ID),]$trisomy19
protCLL$SF3B1 <- patMeta[match(colnames(protCLL), patMeta$Patient.ID),]$SF3B1
protCLL$del11q <- patMeta[match(colnames(protCLL), patMeta$Patient.ID),]$del11q
protSub <- protCLL[,!is.na(protCLL$trisomy19) & (protCLL$trisomy12 == 1) & (protCLL$IGHV.status == "M")]Samples in the test and their trisomy19 status
tri19 <- structure(protSub$trisomy19, names = colnames(protSub))
sort(tri19)P0021 P0038 P0107 P0611 P0735 P0045 P0098 P0494 P0615 P0648
0 0 0 0 0 1 1 1 1 1
Levels: 0 1Detect differentially expressed proteins (proDA)
testMat <- assay(protSub)
fit <- proDA(testMat, design = ~ 1 + tri19)
resTab <- test_diff(fit, contrast = "tri191") %>%
dplyr::rename(id = name, logFC = diff, t=t_statistic,
P.Value = pval, adj.P.Val = adj_pval) %>%
mutate(name = rowData(protSub[id,])$hgnc_symbol) %>%
select(name, id, logFC, t, P.Value, adj.P.Val) %>%
arrange(P.Value) %>%
as_tibble()P-value histogram
hist(resTab$P.Value, breaks = 50)
Table of associations with P Value < 0.05
corRes.sig <- resTab %>% filter(P.Value < 0.01) %>%
mutate(chromosome = rowData(protSub[id,])$chromosome_name)
corRes.sig %>% mutate_if(is.numeric, formatC, digits=2, format="e") %>% DT::datatable()Volcano plot (50% FDR)
plotVolcano <- function(pTab, fdrCut = 0.05, posCol = "red", negCol = "blue",
x_lab = "dm", plotTitle = "",ifLabel = FALSE,
colLabel = NULL) {
plotTab <- pTab %>% mutate(ifSig = ifelse(adj.P.Val > fdrCut, "n.s.",
ifelse(logFC > 0, "up","down"))) %>%
mutate(ifSig = factor(ifSig, levels = c("up","down","n.s.")))
pCut <- -log10((filter(plotTab, ifSig != "n.s.") %>% arrange(desc(P.Value)))$P.Value[1])
g <- ggplot(plotTab, aes(x=logFC, y=-log10(P.Value), label = name)) +
geom_point(shape = 21, aes(fill = ifSig),size=3) +
geom_hline(yintercept = pCut, linetype = "dashed") +
annotate("text", x = -Inf, y = pCut, label = paste0(fdrCut*100,"% FDR"),
size = 5, vjust = -1.2, hjust=-0.1) +
scale_fill_manual(values = c(n.s. = "grey70",
up = posCol, down = negCol)) +
theme( legend.position = "bottom",
legend.text = element_text(size = 15)) +
ylab(expression(-log[10]*'('*italic(P)~value*')')) +
xlab(x_lab) + ggtitle(plotTitle)
if (ifLabel & is.null(colLabel))
g <- g + ggrepel::geom_text_repel(data = filter(plotTab, ifSig != "n.s."),
size=5, force = 2)
else if (ifLabel & !is.null(colLabel)) {
g <- g+ggrepel::geom_text_repel(data = filter(plotTab, ifSig != "n.s."),
aes_string(col = colLabel),
size=5, force = 2) +
scale_color_manual(values = c(yes = "red",no = "black"))
}
return(g)
}
plotTab <- resTab %>%
mutate(chromosome = rowData(protCLL[id,])$chromosome_name) %>%
mutate(onChr19 = ifelse(chromosome == "19","yes","no"))
plotVolcano(plotTab, fdrCut =0.5, x_lab="log2FoldChange",
plotTitle = "trisomy19", ifLabel = TRUE, colLabel = "onChr19")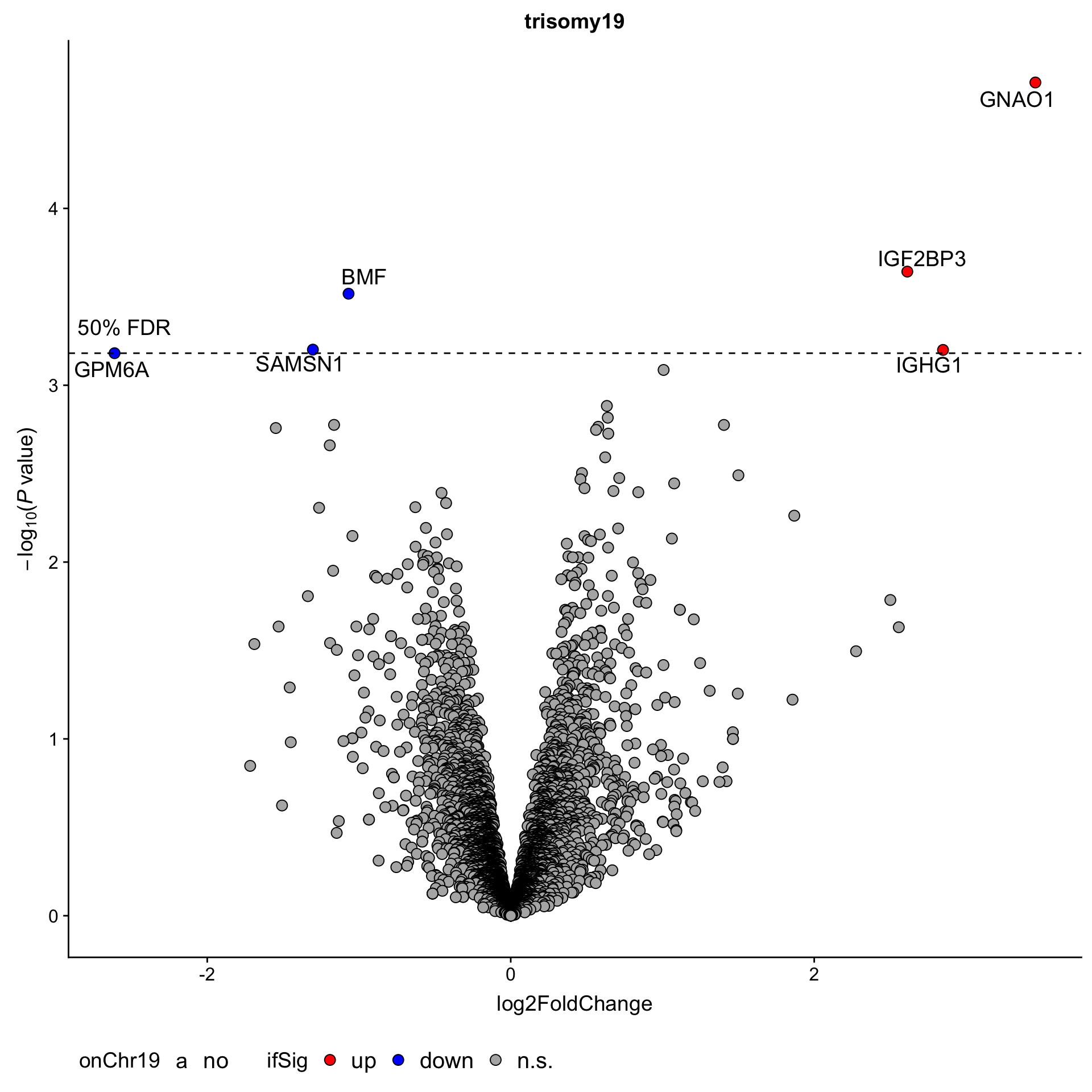
Plot top 9 most differentially expressed proteins
protTab <- sumToTiday(protSub,"patID") %>% mutate(name = hgnc_symbol)
plotTab <- filter(protTab, name %in% corRes.sig$name[1:9]) %>% dplyr::rename(expression = "QRILC")
ggplot(plotTab, aes(x=trisomy19, y = expression)) + geom_boxplot(aes(fill = trisomy19)) + geom_point() +
ggrepel::geom_text_repel(aes(label = colID)) +
facet_wrap(~name, scale = "free")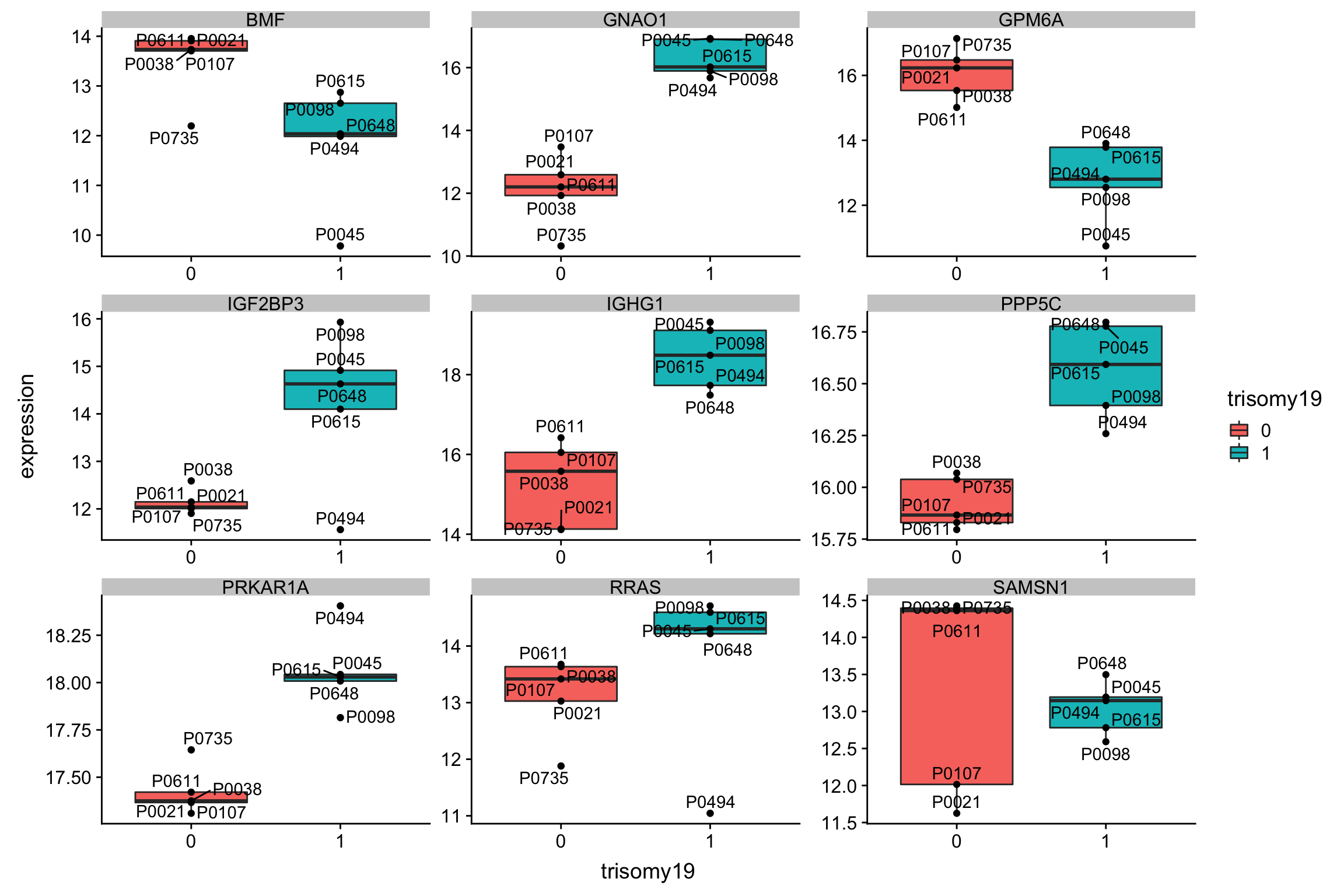 Although the separation looks good, the effect sizes are generally small. This may also lead to high adjusted p-values
Although the separation looks good, the effect sizes are generally small. This may also lead to high adjusted p-values
Heatmap of differentially expressed proteins
proList <- filter(corRes.sig, !is.na(name)) %>% distinct(name, .keep_all = TRUE) %>% pull(id)
plotMat <- assays(protSub)[["QRILC"]][proList,]
rownames(plotMat) <- rowData(protSub[proList,])$hgnc_symbol
colAnno <- colData(protSub)[,c("trisomy19","trisomy12")] %>%
data.frame()
rowAnno <- rowData(protCLL)[proList, c("chromosome_name","hgnc_symbol"),drop=FALSE] %>% data.frame(stringsAsFactors = FALSE) %>%
mutate(onChr19 = ifelse(chromosome_name == "19","yes","no")) %>%
select(hgnc_symbol, onChr19) %>% data.frame() %>% column_to_rownames("hgnc_symbol")
plotMat <- jyluMisc::mscale(plotMat, censor = 6)
pheatmap(plotMat, scale = "none", annotation_col = colAnno, annotation_row = rowAnno,
clustering_method = "ward.D2",
color = colorRampPalette(c("navy","white","firebrick"))(100),
breaks = seq(-6,6, length.out = 101))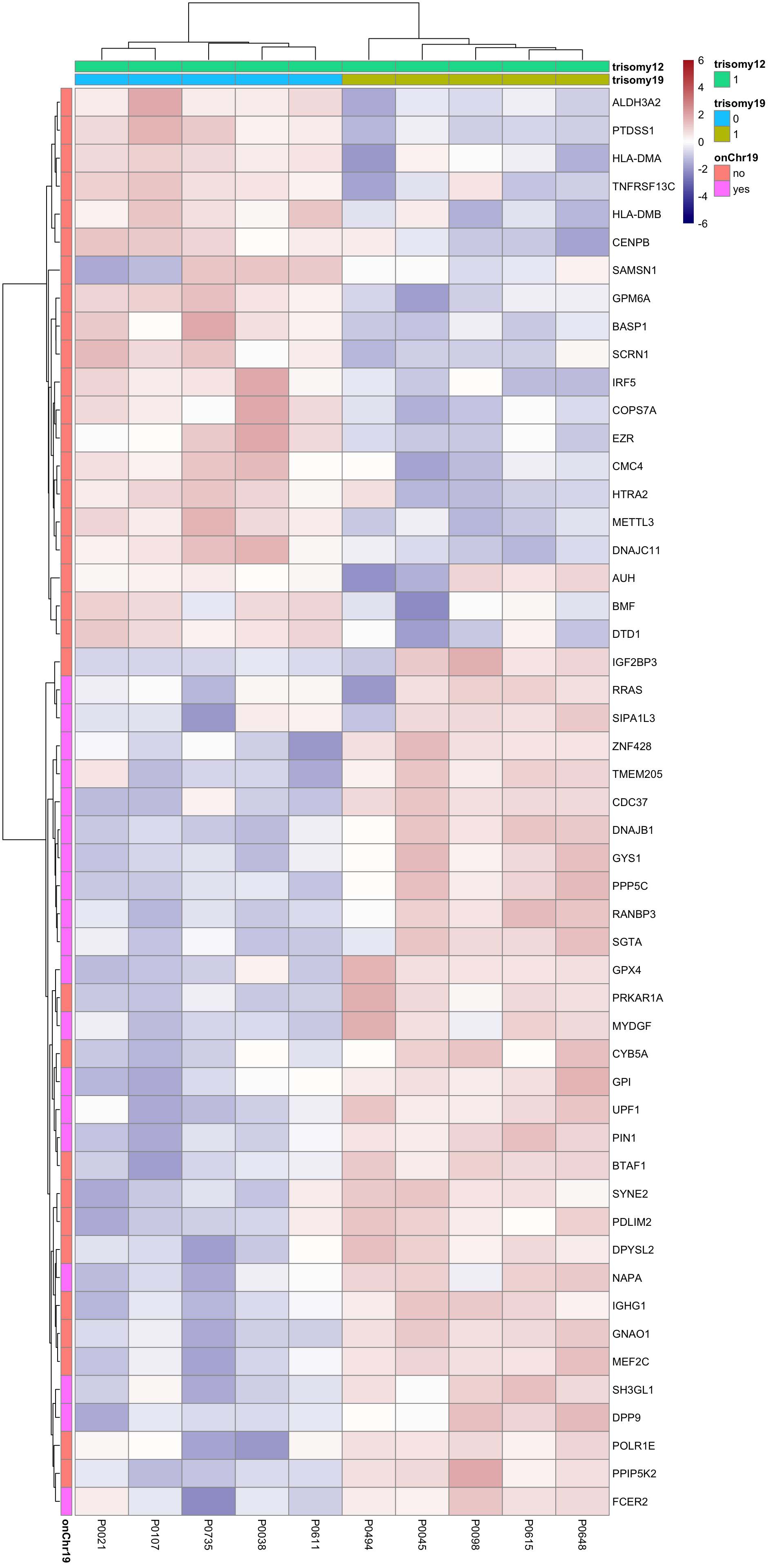 Trisomy19 samples form its own cluster.
Trisomy19 samples form its own cluster.
Enrichment analysis
gmts = list(H= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
inputTab <- resTab %>% filter(P.Value <0.05) %>%
mutate(name = rowData(protCLL[id,])$hgnc_symbol) %>% filter(!is.na(name)) %>%
distinct(name, .keep_all = TRUE) %>%
select(name, t) %>% data.frame() %>% column_to_rownames("name")
enRes <- list()
enRes[["HALLMARK"]] <- runGSEA(inputTab, gmts$H, "page")
enRes[["KEGG"]] <- runGSEA(inputTab, gmts$KEGG, "page")
p <- plotEnrichmentBar(enRes, pCut =0.05, ifFDR= FALSE)
#pdf("tri12Enrich.pdf", height = 15, width = 6)
plot(p)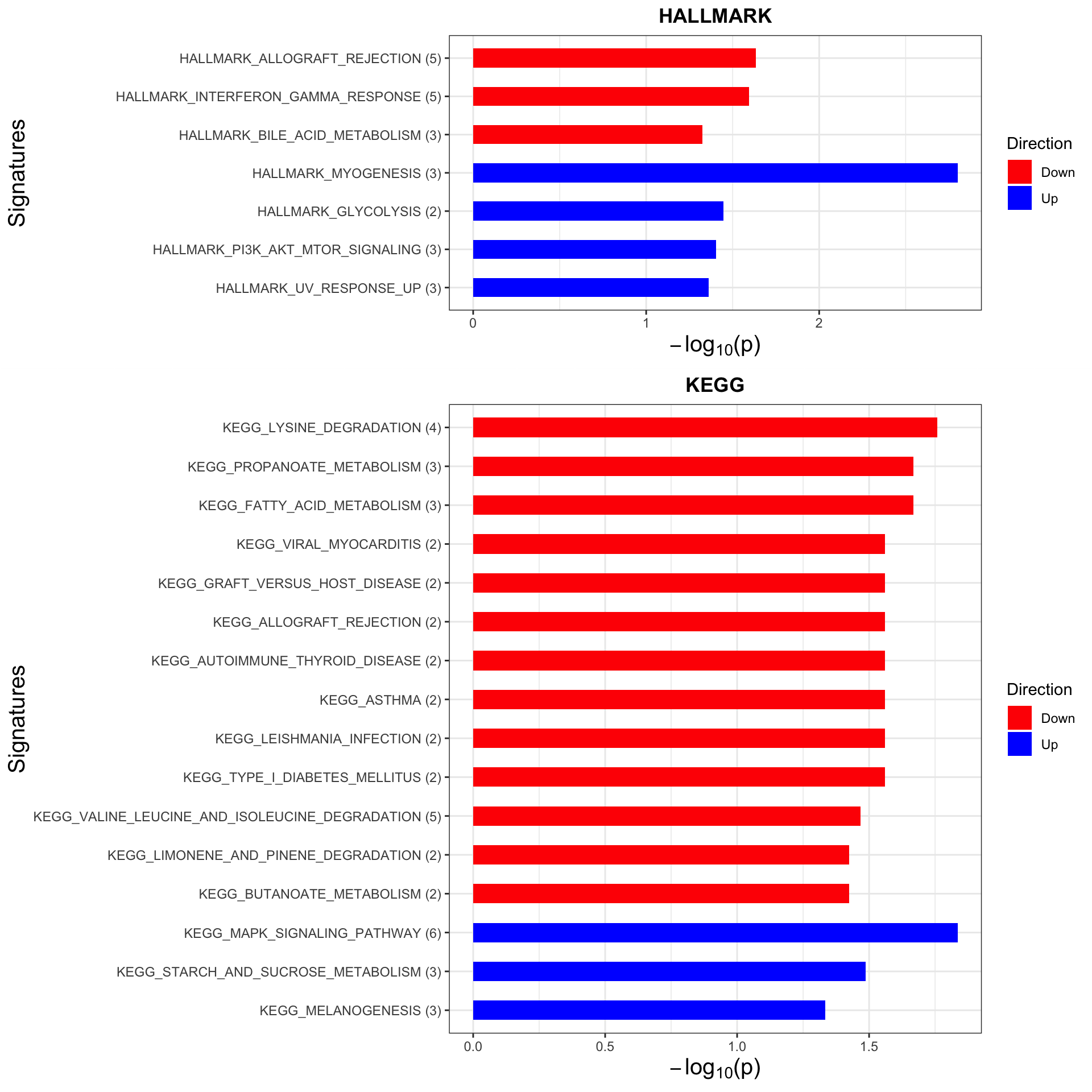
#dev.off()Investigate the impact of trisomy19 on trisomy12 related protein expression in M-CLL
Preprocessing
protCLL$trisomy19 <- patMeta[match(colnames(protCLL), patMeta$Patient.ID),]$trisomy19
protSub <- protCLL[,!is.na(protCLL$trisomy19) & (protCLL$IGHV.status == "M")]
protSub$group <- ifelse(protSub$trisomy12 == 1,
ifelse(protSub$trisomy19 == 1, "both", "onlyTri12"),"none")Distribution of samples
table(protSub$group)
both none onlyTri12
5 14 5 For proteins associated with trisomy12 (10%), test the impact of trisomy19 on them.
protTest <- protSub[resTab.tri12.sig$id,protSub$group != "none"]
testMat <- assay(protTest)
group <- factor(protTest$group, levels = c("both","onlyTri12"))
fit <- proDA(testMat, design = ~ group)
resTab.tri19 <- test_diff(fit, contrast = "grouponlyTri12") %>%
dplyr::rename(id = name, logFC = diff, t=t_statistic,
P.Value = pval, adj.P.Val = adj_pval) %>%
mutate(name = rowData(protTest[id,])$hgnc_symbol) %>%
select(name, id, logFC, t, P.Value, adj.P.Val,n_obs) %>%
arrange(P.Value) %>%
as_tibble()Compare the two results
resList <- list()
resList[["tri12_up"]] <- filter(resTab.tri12.sig, logFC >0)$name
resList[["tri12_down"]] <- filter(resTab.tri12.sig, logFC <0)$name
resList[["tri19_up"]] <- filter(resTab.tri19, P.Value <= 0.01, logFC >0)$name
resList[["tri19_down"]] <- filter(resTab.tri19, P.Value <= 0.01, logFC <0)$name
upset(fromList(resList))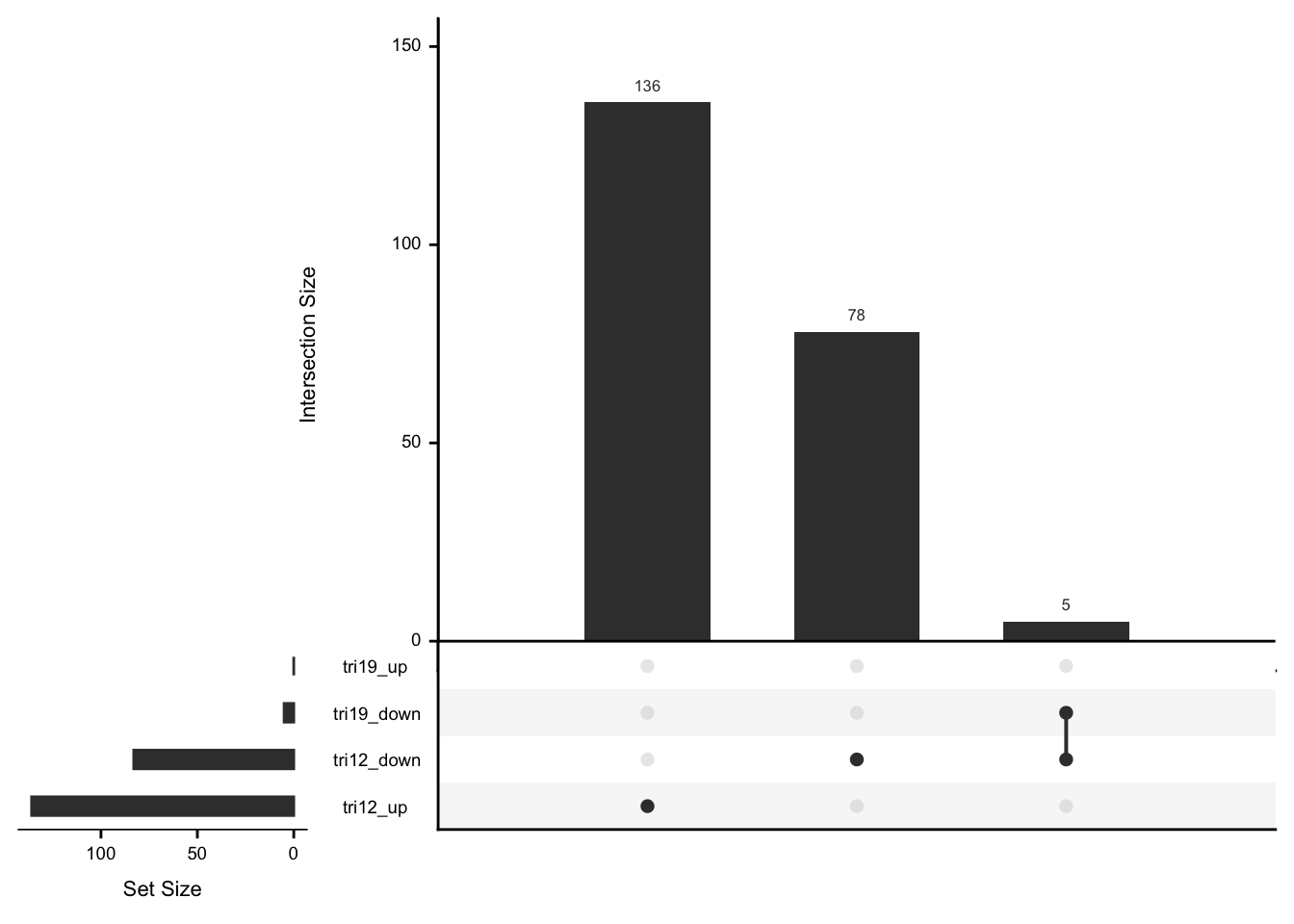
Plot the expressions of proteins that show additional change in trisomy19 samples (raw P-value < 0.01)
proteinList <- intersect(filter(resTab.tri19, P.Value <=0.01)$name, resTab.tri12.sig$name)
protTab <- sumToTiday(protSub,"patID") %>% mutate(name = hgnc_symbol)
plotList <- lapply(proteinList, function(symbol) {
plotTab <- filter(protTab, name == symbol) %>%
mutate(group = factor(group, levels = c("none","onlyTri12","both")))
chrLoc <- unique(filter(protTab, hgnc_symbol == symbol)$chromosome_name)
ggplot(plotTab, aes(x=group, y = count)) + geom_boxplot(aes(fill = group)) + geom_point() +
ggtitle(sprintf("%s (on chr%s)",symbol, chrLoc)) +
ggrepel::geom_text_repel(aes(label = colID))
})
plot_grid(plotlist= plotList, ncol =3)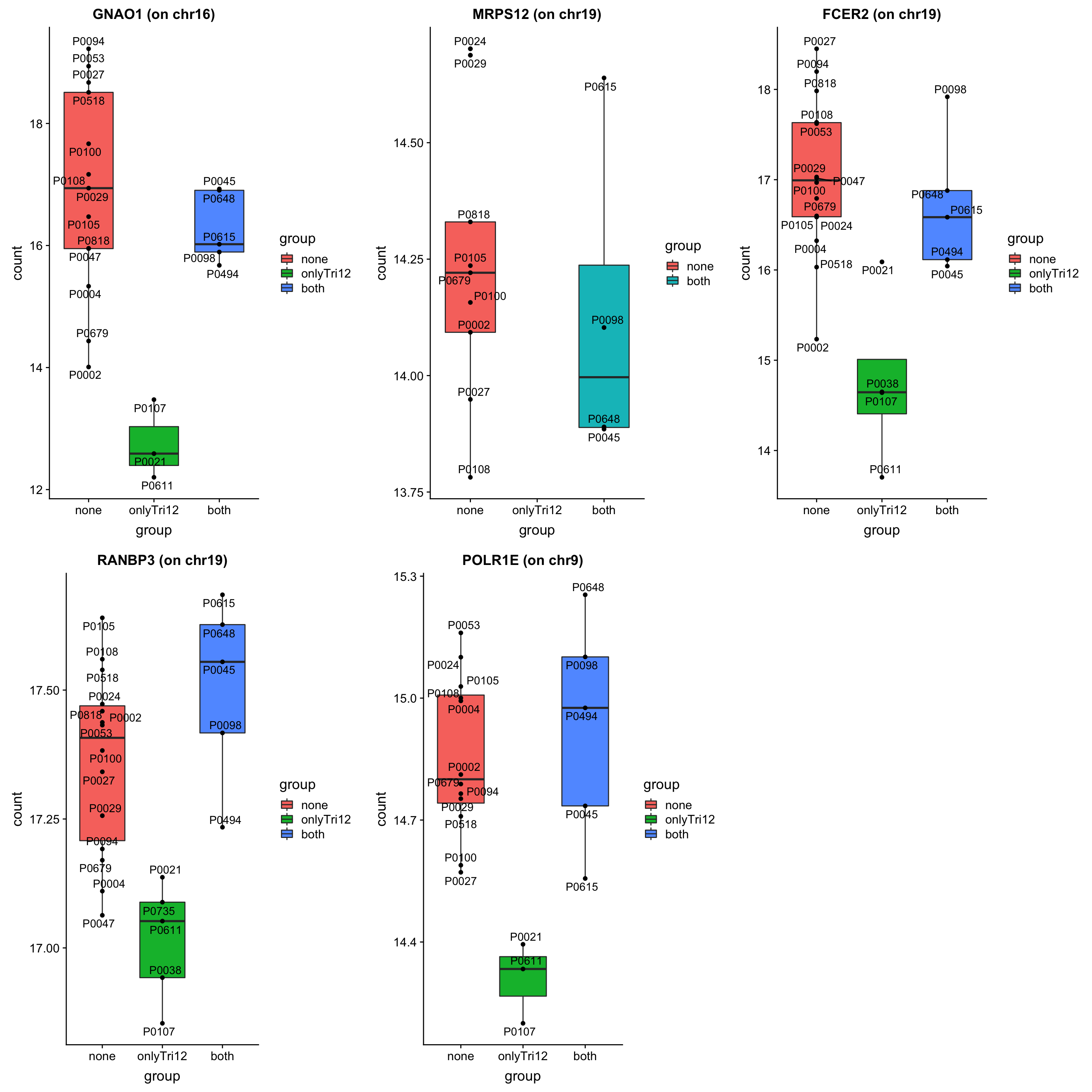 As we don’t have samples with only trisomy19 but not trisomy12, we don’t have a signature of “pure” trisomy19 effect on expression. So those are not “epistatic interactions”. But can be considered as additional effect of trisomy19 on the genes related to trisomy12.
As we don’t have samples with only trisomy19 but not trisomy12, we don’t have a signature of “pure” trisomy19 effect on expression. So those are not “epistatic interactions”. But can be considered as additional effect of trisomy19 on the genes related to trisomy12.
Transcriptomic analysis
Preprocessing data
Sample subsetting
Subset sample: select M-CLLs with annotation for trisomy12 and trisomy19
dds$IGHV <- patMeta[match(colnames(dds),patMeta$Patient.ID),]$IGHV.status
dds$trisomy12 <- patMeta[match(colnames(dds),patMeta$Patient.ID),]$trisomy12
dds$trisomy19 <- patMeta[match(colnames(dds),patMeta$Patient.ID),]$trisomy19
ddsSub <- dds[,dds$diag == "CLL" & dds$IGHV %in% "M" & !is.na(dds$trisomy12) & !is.na(dds$trisomy19)]Summary of trisomy12 and trisomy19 status
annoTab <- tibble(patID = colnames(ddsSub),
tri12 = ifelse(ddsSub$trisomy12 == 1, "tri12","wt"),
tri19 = ifelse(ddsSub$trisomy19 ==1, "tri19","wt")) %>%
mutate(group = ifelse(tri12 == "tri12", ifelse(tri19 == "tri19","both","onlyTri12"),"none"))
table(annoTab$tri12, annoTab$tri19)
tri19 wt
tri12 5 7
wt 0 97There are too many samples without either trisomy19 and trisomy12. The sample imbalance will cause bias in hypothesis testing. In addition, the other recurrent alterations in wildtype group may also complicate the problem.
To ensure the compatibility and reduce noise, in the below analysis, I will:
Check the distribution of several recurrent mutations that impact gene expression (based on Almut’s paper), namely: del13q, SF3B1, del17p, TP53, del11q, BRAF, gain8q, del8p, NOTCH1, MED12, ATM.
Get mutations that occurred in non-wildtype samples (with trisomy12, trisomy19 or both).
Remove wild type samples that contain the recurrent mutations that do not occur in non-wildtype samples.
Block for the mutations that occur in both wild type and non-wildtype samples.
#Get the distribution of recurrent mutations in RNAseq samples
patSum <- select(patMeta, Patient.ID, del13q, SF3B1, del17p, TP53, del11q, BRAF, gain8q, del8p, NOTCH1, MED12, ATM) %>%
filter(Patient.ID %in% annoTab$patID) %>%
gather(key = "gene",value = "status",-Patient.ID) %>%
mutate(status = as.integer(status)) %>% filter(status %in% 1)Which mutations occur in non-wildtype samples?
geneKeep <- unique(filter(patSum, Patient.ID %in% filter(annoTab, group != "none")$patID)$gene)
geneKeep[1] "del13q" "SF3B1" "del17p" "TP53" Remove wildtype samples with recurrent mutations except for those four: del13q, SF3B1, del17p and TP53. Those genes will be blocked when doing hypothesis test
patSum.other <- filter(patSum, !gene %in% geneKeep)
annoTab <- filter(annoTab, !(group == "none" & patID %in% patSum.other$Patient.ID))Sample summary after filtering
table(annoTab$tri12, annoTab$tri19)
tri19 wt
tri12 5 7
wt 0 82Filtering transcripts
ddsSub <- ddsSub[,annoTab$patID]
ddsSub$group <- annoTab$group
geneTab <- patMeta[match(ddsSub$PatID, patMeta$Patient.ID),c("Patient.ID", "del13q","SF3B1","del17p","TP53")] %>%
data.frame() %>% column_to_rownames("Patient.ID")
colData(ddsSub) <- cbind(colData(ddsSub),geneTab)
#filter out none protein coding genes and gene on sex chromosome
ddsSub<-ddsSub[rowData(ddsSub)$biotype %in% "protein_coding",]
ddsSub <- ddsSub[! rowData(ddsSub)$symbol %in% c("",NA),]
##vst
ddsSub.vst <- varianceStabilizingTransformation(ddsSub)
dim(ddsSub)[1] 20074 94Differentially expressed RNAs related to trisomy19 in M-CLL samples with trisomy12
Preprocessing
ddsTest <- ddsSub[, ddsSub$group %in% c("onlyTri12","both")]
##filter out low count genes
minrs <- 100
rs <- rowSums(counts(ddsTest, normalized = TRUE))
ddsTest<-ddsTest[ rs >= minrs, ]Detect differentially expressed genes using DESeq2
design(ddsTest) <- ~ del13q + SF3B1 + del17p + TP53 + trisomy19
#design(ddsTest) <- ~trisomy19
ddsTest <- DESeq(ddsTest)using pre-existing size factorsestimating dispersionsgene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesfitting model and testingresTab.rna <- results(ddsTest, contrast = c("trisomy19","1","0"), tidy = TRUE) %>%
dplyr::rename(logFC = log2FoldChange, P.Value = pvalue, adj.P.Val = padj, id=row) %>%
mutate(chromosome = rowData(dds[id,])$chromosome,
name = rowData(dds[id,])$symbol) %>%
arrange(P.Value)P-value histogram
hist(resTab.rna$P.Value, breaks = 50)
Table of associations with 10% FDR
corRes.sig <- resTab.rna %>% filter(adj.P.Val <0.1)
corRes.sig %>% select(id, name, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=2, format="e") %>% DT::datatable()Volcano plot (1% FDR)
plotTab <- resTab.rna %>%
mutate(onChr19 = ifelse(chromosome == "19","yes","no"))
plotVolcano(plotTab, fdrCut =0.01, x_lab="log2FoldChange",
plotTitle = "trisomy19", ifLabel = TRUE, colLabel = "onChr19")Warning: Removed 50 rows containing missing values (geom_point).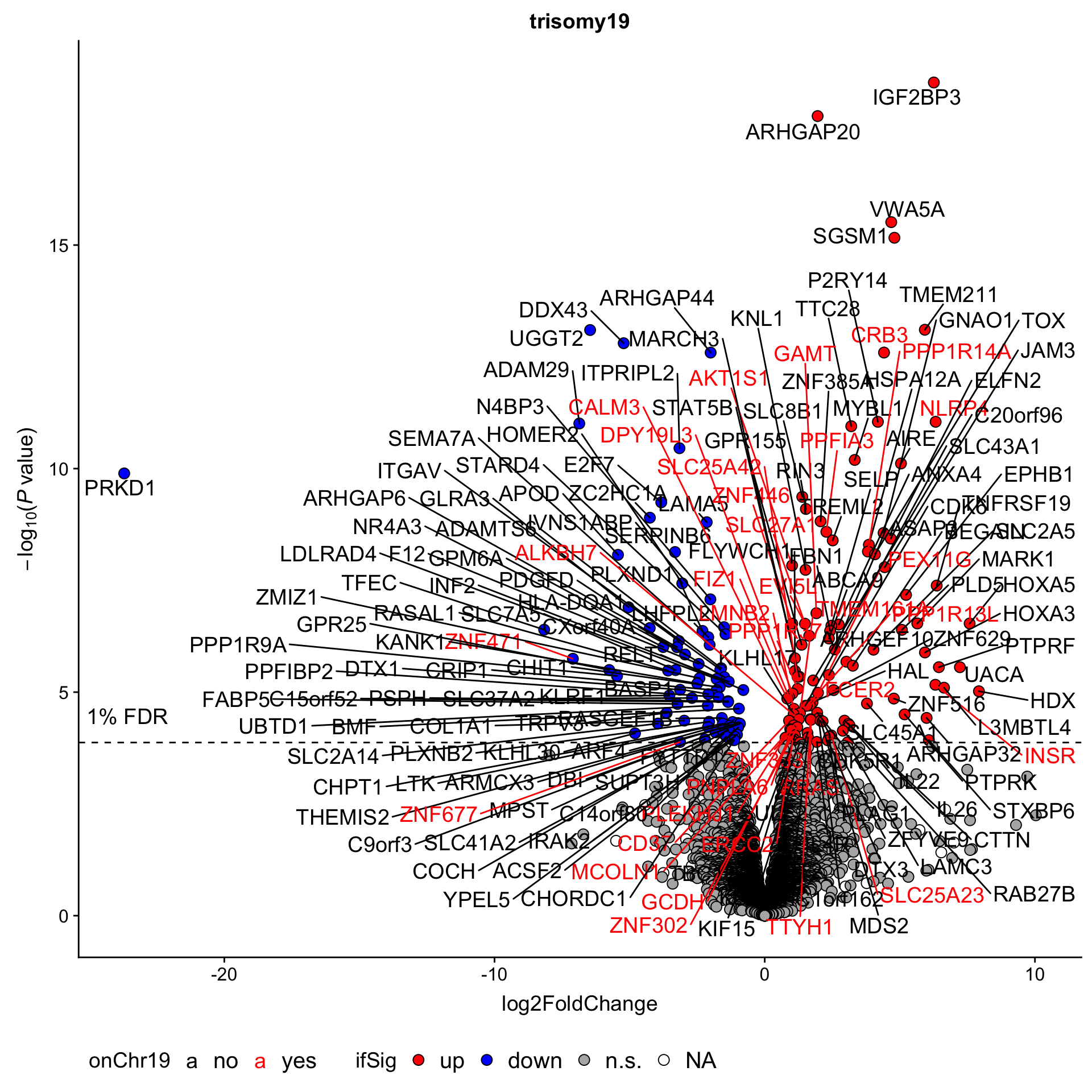
Plot top 9 most differentially expressed proteins
exprTab <- assay(ddsSub.vst[corRes.sig$id[1:9],colnames(ddsTest)]) %>% data.frame() %>%
rownames_to_column("id") %>%
mutate(name = rowData(ddsSub[id])$symbol) %>%
gather(key = "patID", value = "expression", -id, -name) %>%
mutate(trisomy19 = ddsSub[,patID]$trisomy19)
plotTab <- filter(exprTab, name %in% corRes.sig$name[1:9])
ggplot(plotTab, aes(x=trisomy19, y = expression)) + geom_boxplot(aes(fill = trisomy19)) + geom_point() +
facet_wrap(~name, scale = "free") +
ggrepel::geom_text_repel(aes(label = patID))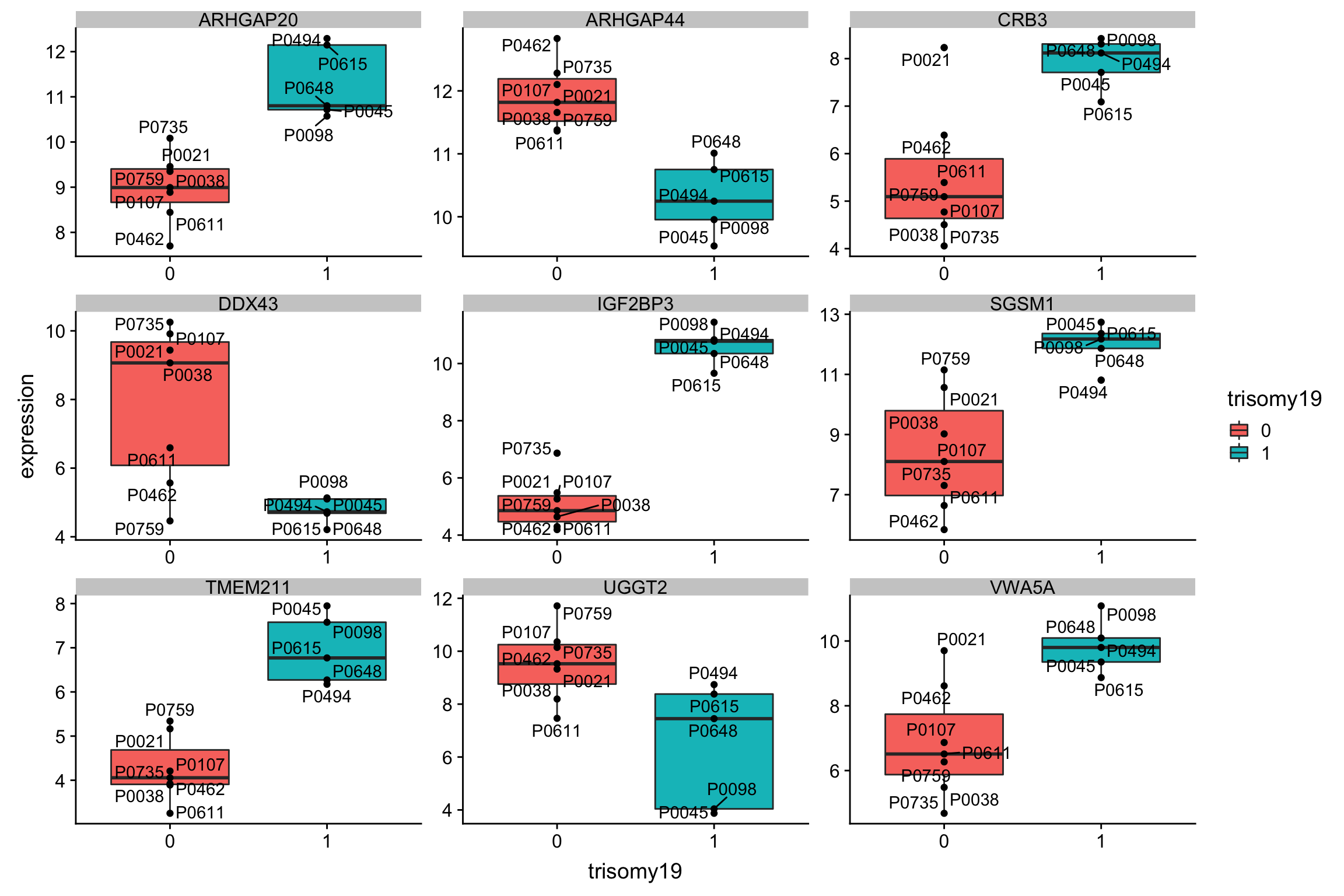
Heatmap of differentially expressed protein (1% FDR)
proList <- filter(corRes.sig, adj.P.Val < 0.01) %>% arrange(desc(logFC)) %>%
filter(!is.na(name)) %>% distinct(name, .keep_all = TRUE) %>% pull(id)
plotMat <- assay(ddsSub.vst)[proList,colnames(ddsTest)]
rownames(plotMat) <- rowData(ddsSub.vst[proList,])$symbol
colAnno <- colData(ddsSub.vst)[,c("trisomy19","trisomy12","del13q","SF3B1","TP53", "del17p")] %>%
data.frame()
rowAnno <- filter(corRes.sig, adj.P.Val < 0.01) %>%
mutate(onChr19 = ifelse(chromosome == "19","yes","no")) %>%
select(name, onChr19) %>% data.frame() %>% column_to_rownames("name")
plotMat <- jyluMisc::mscale(plotMat, censor = 6)
pheatmap(plotMat, scale = "none", annotation_col = colAnno, annotation_row = rowAnno, cluster_rows = FALSE,
clustering_method = "ward.D2",
color = colorRampPalette(c("navy","white","firebrick"))(100),
breaks = seq(-6,6, length.out = 101))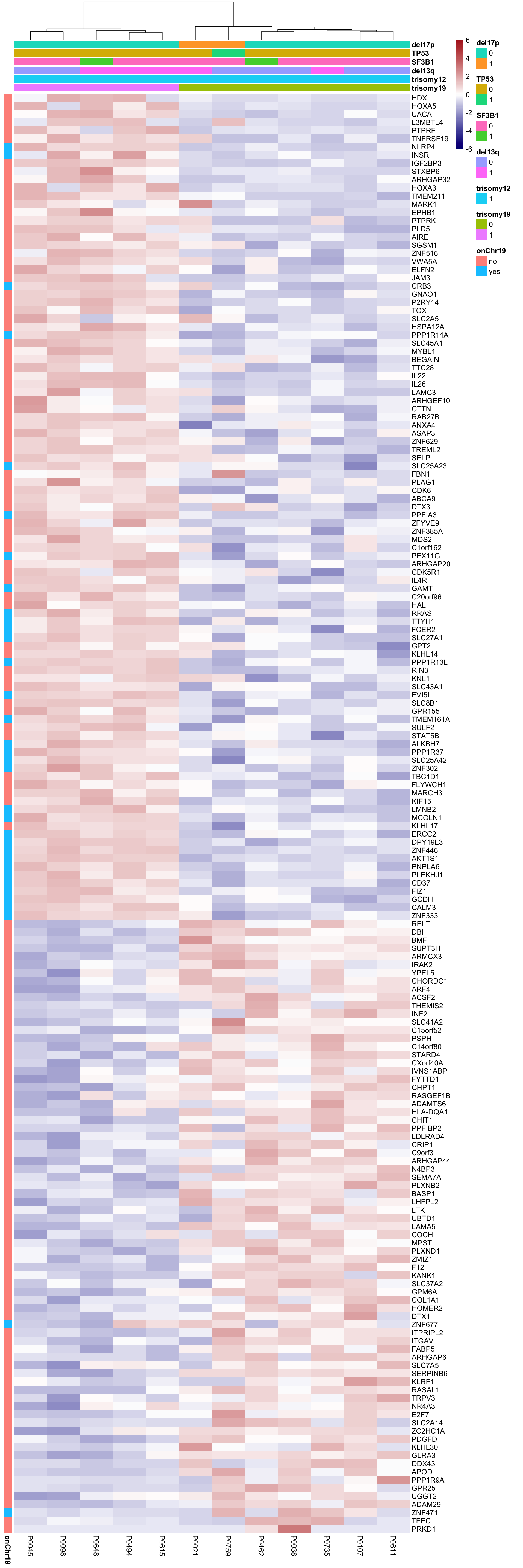 Trisomy19 samples form its own cluster.
Trisomy19 samples form its own cluster.
Enrichment analysis (10% FDR)
gmts = list(H= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
inputTab <- resTab.rna %>% filter(P.Value <0.05) %>%
distinct(name, .keep_all = TRUE) %>%
select(name, stat) %>% data.frame() %>% column_to_rownames("name")
enRes <- list()
enRes[["HALLMARK"]] <- runGSEA(inputTab, gmts$H, "page")
enRes[["KEGG"]] <- runGSEA(inputTab, gmts$KEGG, "page")
p <- plotEnrichmentBar(enRes, pCut =0.1, ifFDR= TRUE)
#pdf("tri12Enrich.pdf", height = 15, width = 6)
plot(p)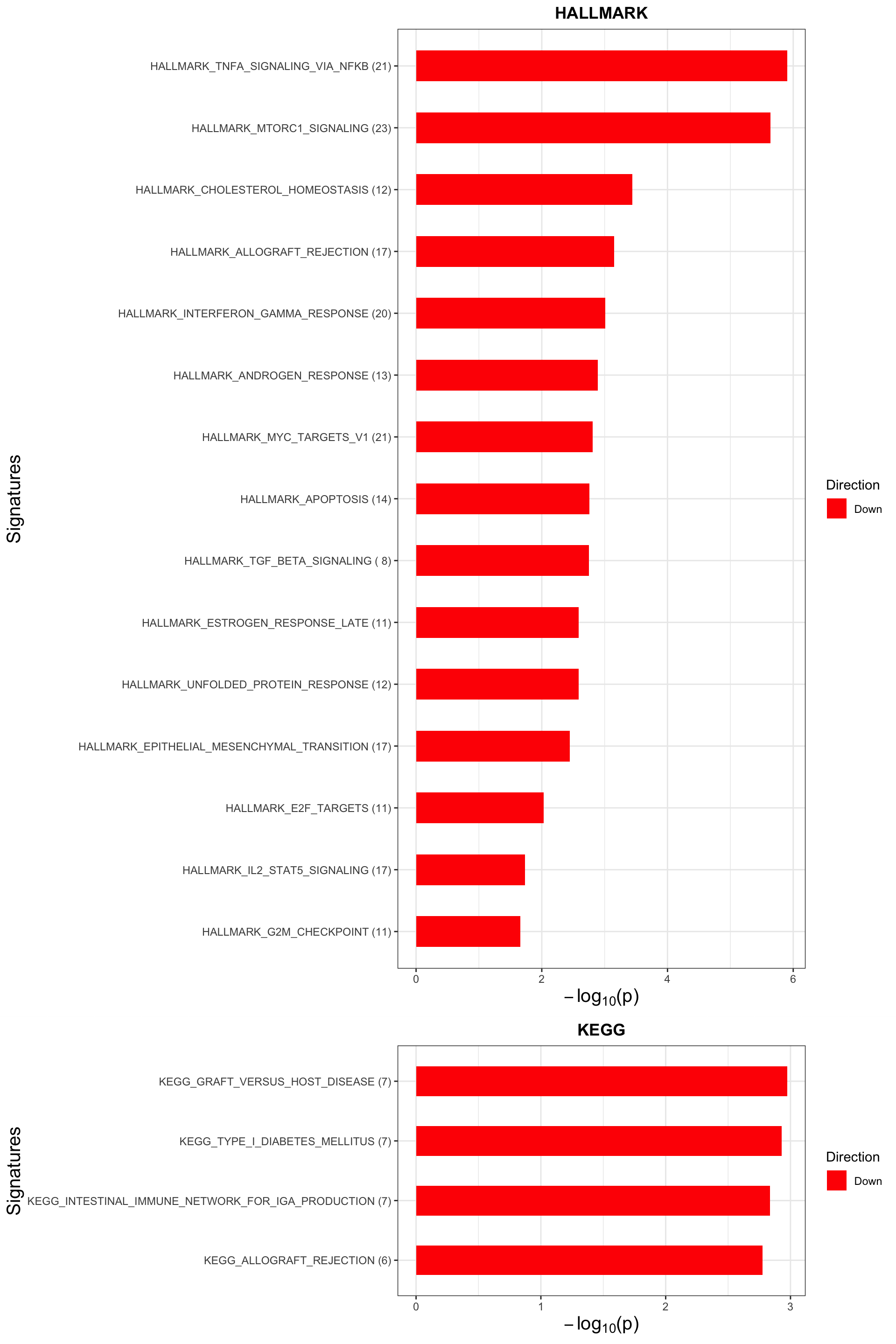
#dev.off()Heatmap plots for enriched pathways (genes with raw P <0.05)
HALLMARK_TNFA_SIGNALING_VIA_NFKB
plotSetHeatmap <- function(geneSigTab, setDir, setName, ddsObj, colAnno, scale = TRUE, annoChrom = NULL) {
geneList <- loadGSC(setDir)[["gsc"]][[setName]]
sigGene <- filter(geneSigTab, name %in% geneList) %>%
arrange(desc(logFC))
colAnno <- colAnno[order(colAnno[,1]),,drop = FALSE]
plotMat <- assay(ddsObj)[sigGene[["id"]],rownames(colAnno)]
if (scale) {
#calculate z-score and sensor
plotMat <- t(scale(t(plotMat)))
plotMat[plotMat >= 6] <- 6
plotMat[plotMat <= -6] <- -6
}
if (!is.null(annoChrom)) {
rowAnno <- mutate(geneSigTab, onChr = ifelse(chromosome %in% annoChrom, "yes","no")) %>%
select(id, onChr) %>% data.frame() %>% column_to_rownames("id")
colnames(rowAnno) <- paste0("onChr",annoChrom)
} else rowAnno <- NULL
pheatmap(plotMat, color = colorRampPalette(c("navy","white","firebrick"))(100),
cluster_cols = FALSE, cluster_rows = FALSE,
annotation_col = colAnno, labels_row = sigGene$name, annotation_row = rowAnno,
show_colnames = TRUE, fontsize_row = 8, breaks = seq(-6,6, length.out = 101), treeheight_row = 0,
border_color = NA, main = setName)
}
colAnnoSub <- colAnno[colnames(ddsTest),c("trisomy19"),drop=FALSE]
resTab.rna.sig <- filter(resTab.rna, P.Value < 0.05)
plotSetHeatmap(resTab.rna.sig, gmts$H, "HALLMARK_TNFA_SIGNALING_VIA_NFKB", ddsSub.vst[,colnames(ddsTest)],
colAnno = colAnnoSub, annoChrom = "19")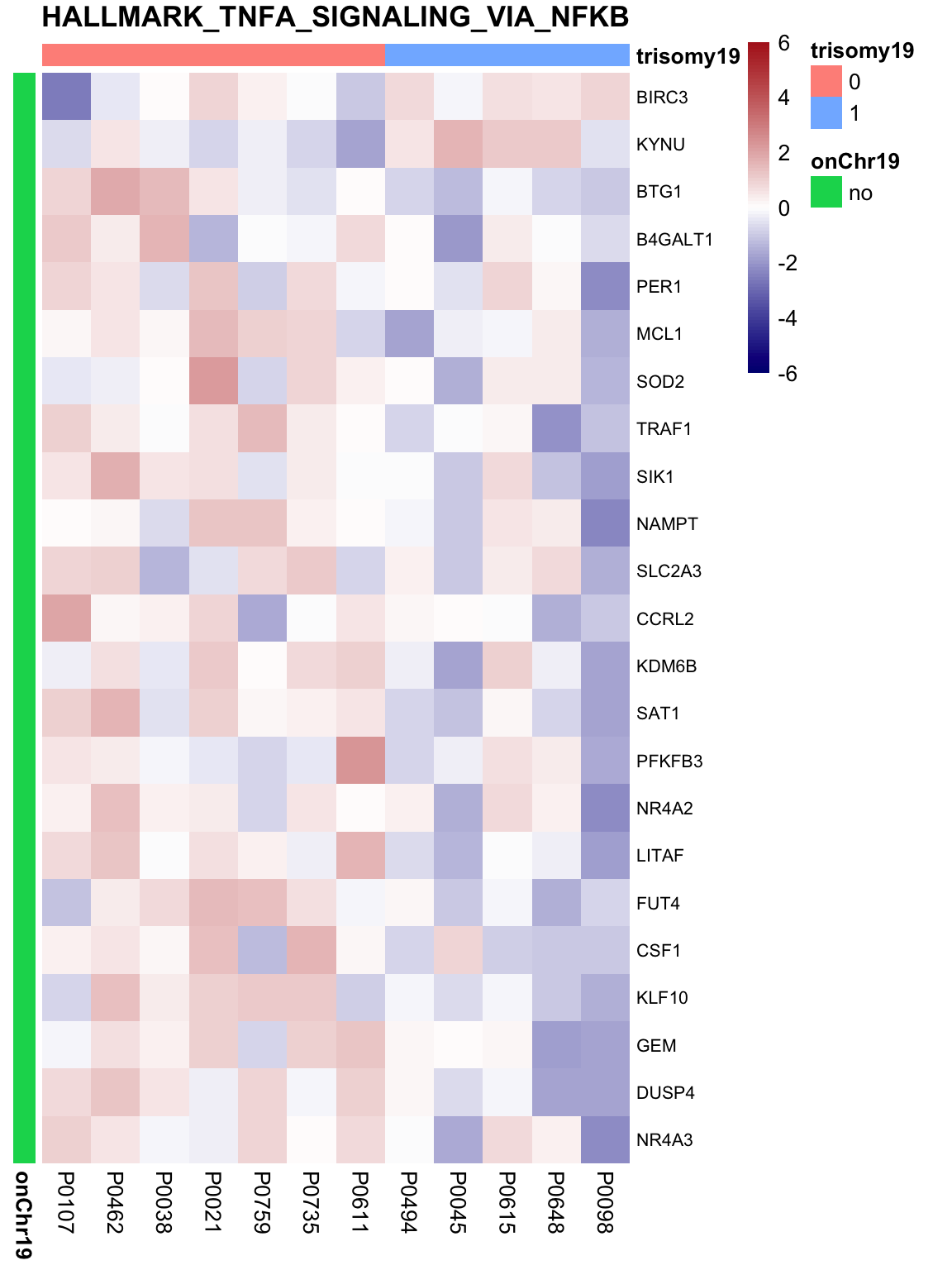
HALLMARK_MTORC1_SIGNALING
plotSetHeatmap(resTab.rna.sig, gmts$H, "HALLMARK_MTORC1_SIGNALING", ddsSub.vst[,colnames(ddsTest)], colAnno = colAnnoSub, annoChrom = "19")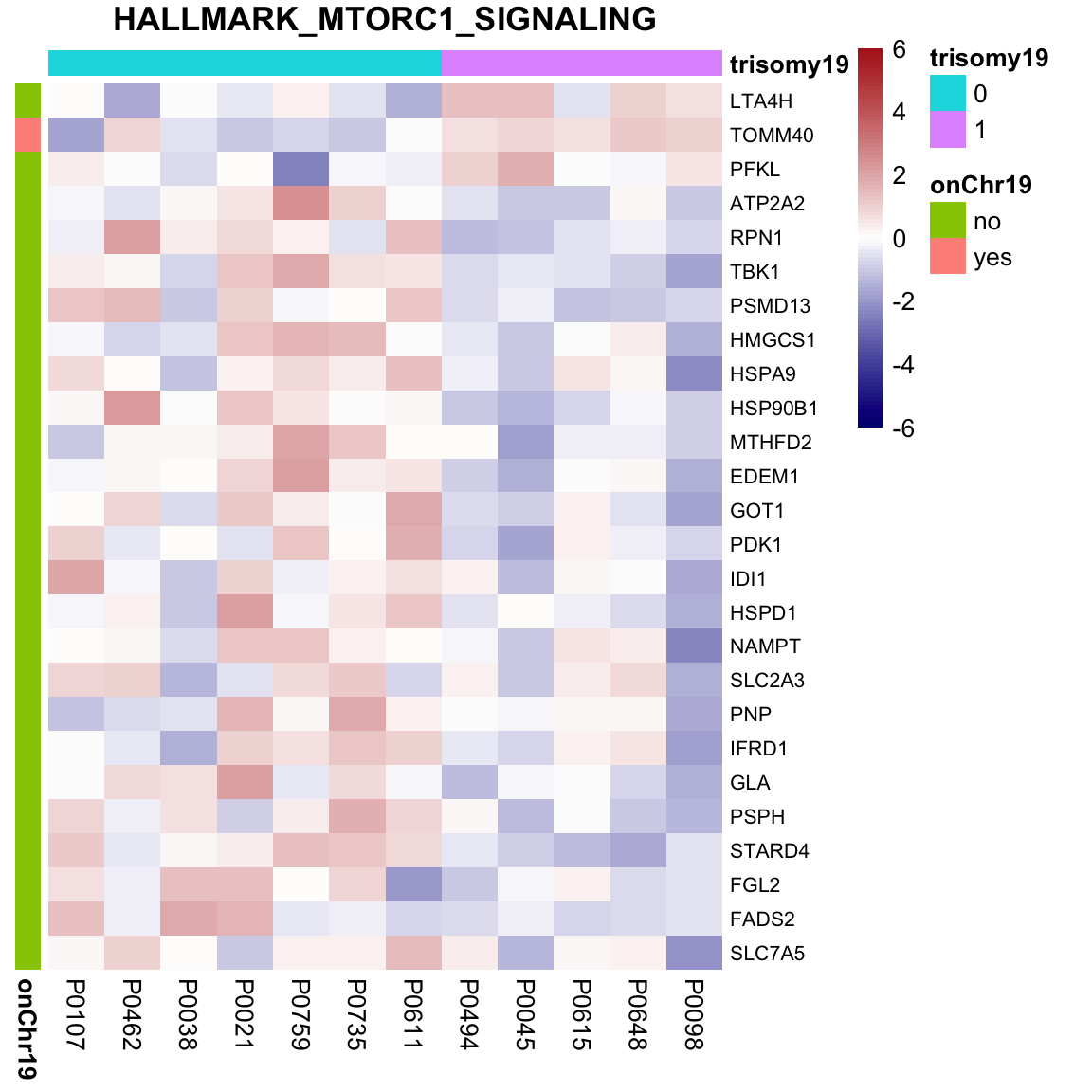
HALLMARK_ALLOGRAFT_REJECTION
plotSetHeatmap(resTab.rna.sig, gmts$H, "HALLMARK_ALLOGRAFT_REJECTION", ddsSub.vst[,colnames(ddsTest)], colAnno = colAnnoSub, annoChrom = "19")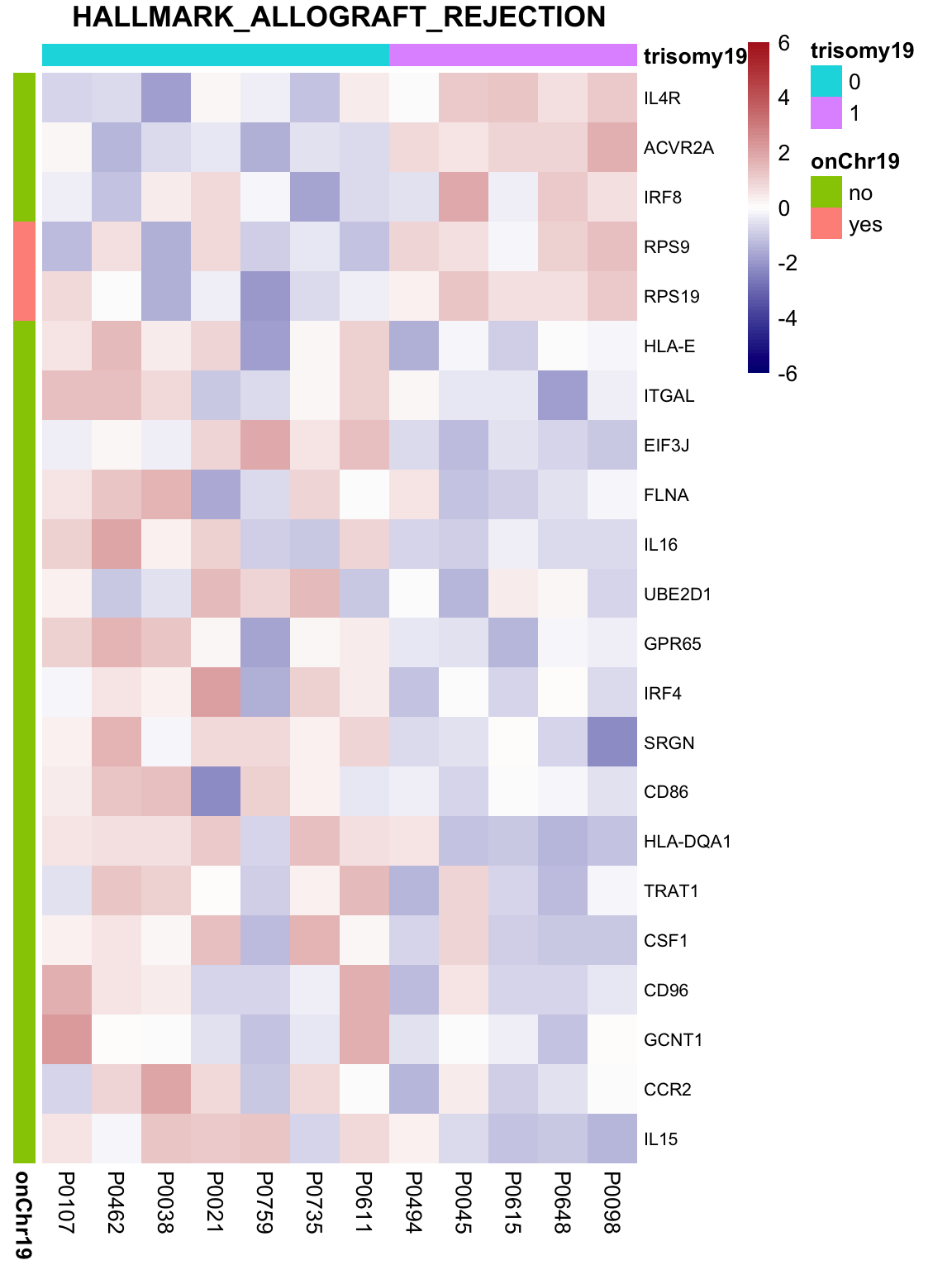
HALLMARK_INTERFERON_GAMMA_RESPONSE
plotSetHeatmap(resTab.rna.sig, gmts$H, "HALLMARK_INTERFERON_GAMMA_RESPONSE", ddsSub.vst[,colnames(ddsTest)], colAnno = colAnnoSub, annoChrom = 19)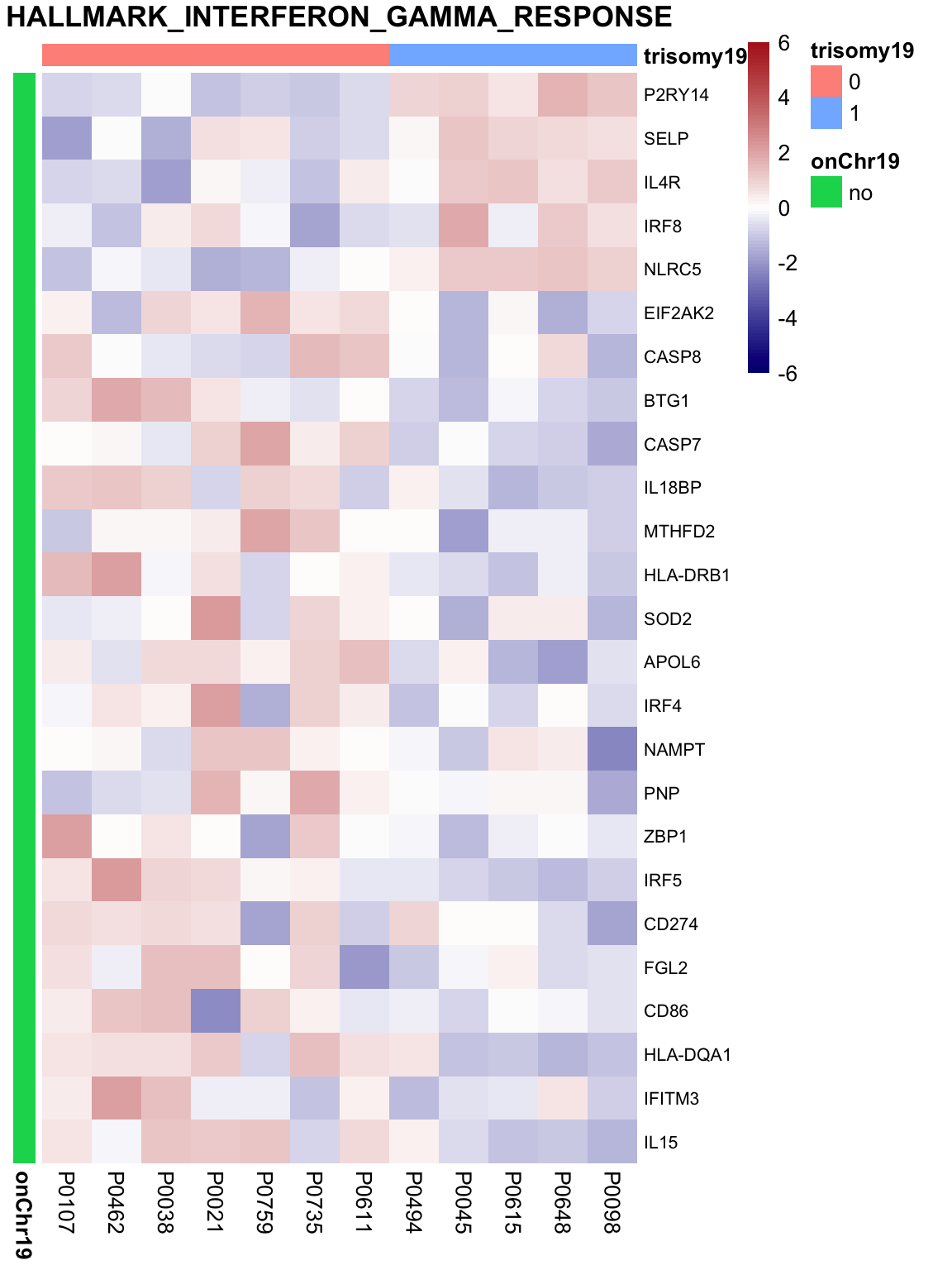
HALLMARK_APOPTOSIS
plotSetHeatmap(resTab.rna.sig, gmts$H, "HALLMARK_APOPTOSIS", ddsSub.vst[,colnames(ddsTest)],
colAnno = colAnnoSub, annoChrom = "19")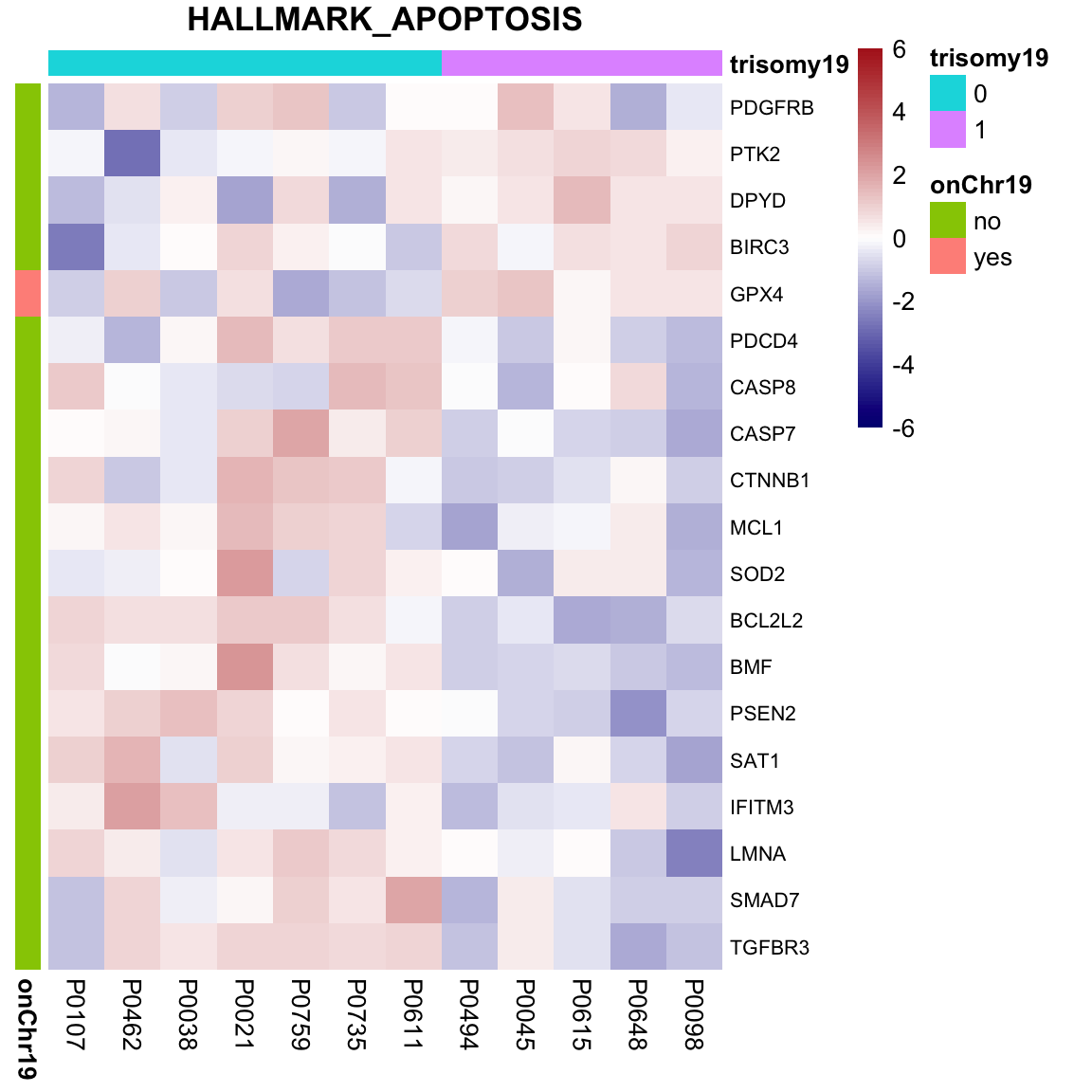
Compare the results with proteomic analysis
Upset plot (I use cut-off of raw p value < 0.01 here)
compareList <- list(Protein_up = filter(resTab, P.Value < 0.01, logFC >0)$name,
Protein_down = filter(resTab, P.Value < 0.01, logFC <0)$name,
RNA_up = filter(resTab.rna, P.Value < 0.01, logFC >0)$name,
RNA_down = filter(resTab.rna, P.Value < 0.01, logFC <0)$name)
upset(fromList(compareList))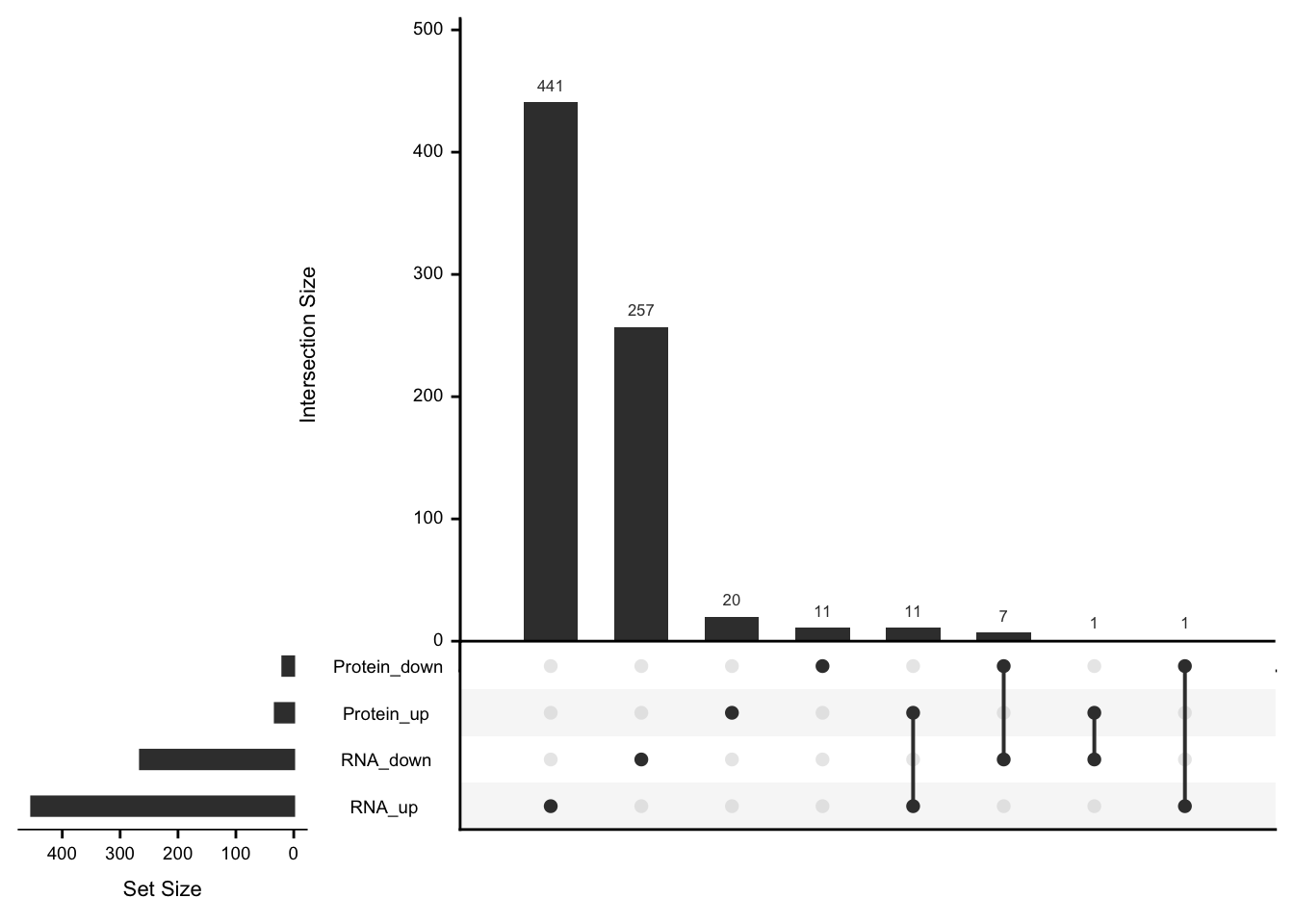
List of genes that show consistant changes at both RNA and protein level
UP in both
intersect(compareList$Protein_up, compareList$RNA_up) [1] "GNAO1" "IGF2BP3" "RRAS" "MEF2C" "CYB5A" "SYNE2" "RANBP3"
[8] "TMEM205" "FCER2" "DPP9" "GPX4" Down in both
intersect(compareList$Protein_down, compareList$RNA_down)[1] "BMF" "SAMSN1" "GPM6A" "ALDH3A2" "BASP1" "IRF5" "EZR" Investigate the impact of trisomy19 on trisomy12 related gene expression in M-CLL
Distribution of samples
table(ddsSub$group)
both none onlyTri12
5 82 7 For proteins associated with trisomy12 (10%), test the impact of trisomy19 on them.
#trisomy19+trisomy12 signature
ddsTest <- ddsSub[,ddsSub$group != "none"]
ddsTest$group <- factor(ddsTest$group, levels = c("onlyTri12","both"))
##filter out low count genes
minrs <- 100
rs <- rowSums(counts(ddsTest, normalized = TRUE))
ddsTest<-ddsTest[ rs >= minrs, ]
design(ddsTest) <- ~ del13q + SF3B1 + del17p + TP53 + group
ddsTest <- DESeq(ddsTest)using pre-existing size factorsestimating dispersionsgene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesfitting model and testingresTab.tri19 <- results(ddsTest, contrast = c("group","both","onlyTri12"), tidy = TRUE) %>%
dplyr::rename(id = row, logFC = log2FoldChange, t=stat,
P.Value = pvalue, adj.P.Val = padj) %>%
mutate(name = rowData(dds[id,])$symbol) %>%
select(name, id, logFC, t, P.Value, adj.P.Val) %>%
arrange(P.Value) %>%
as_tibble()
resTab.tri19.sig <- filter(resTab.tri19, adj.P.Val < 0.1)Compare the two results
resList <- list()
resList[["tri12_up"]] <- filter(resTab.tri12.sig, logFC >0)$name
resList[["tri12_down"]] <- filter(resTab.tri12.sig, logFC <0)$name
resList[["tri19_up"]] <- filter(resTab.tri19.sig, logFC >0)$name
resList[["tri19_down"]] <- filter(resTab.tri19.sig, logFC <0)$name
upset(fromList(resList))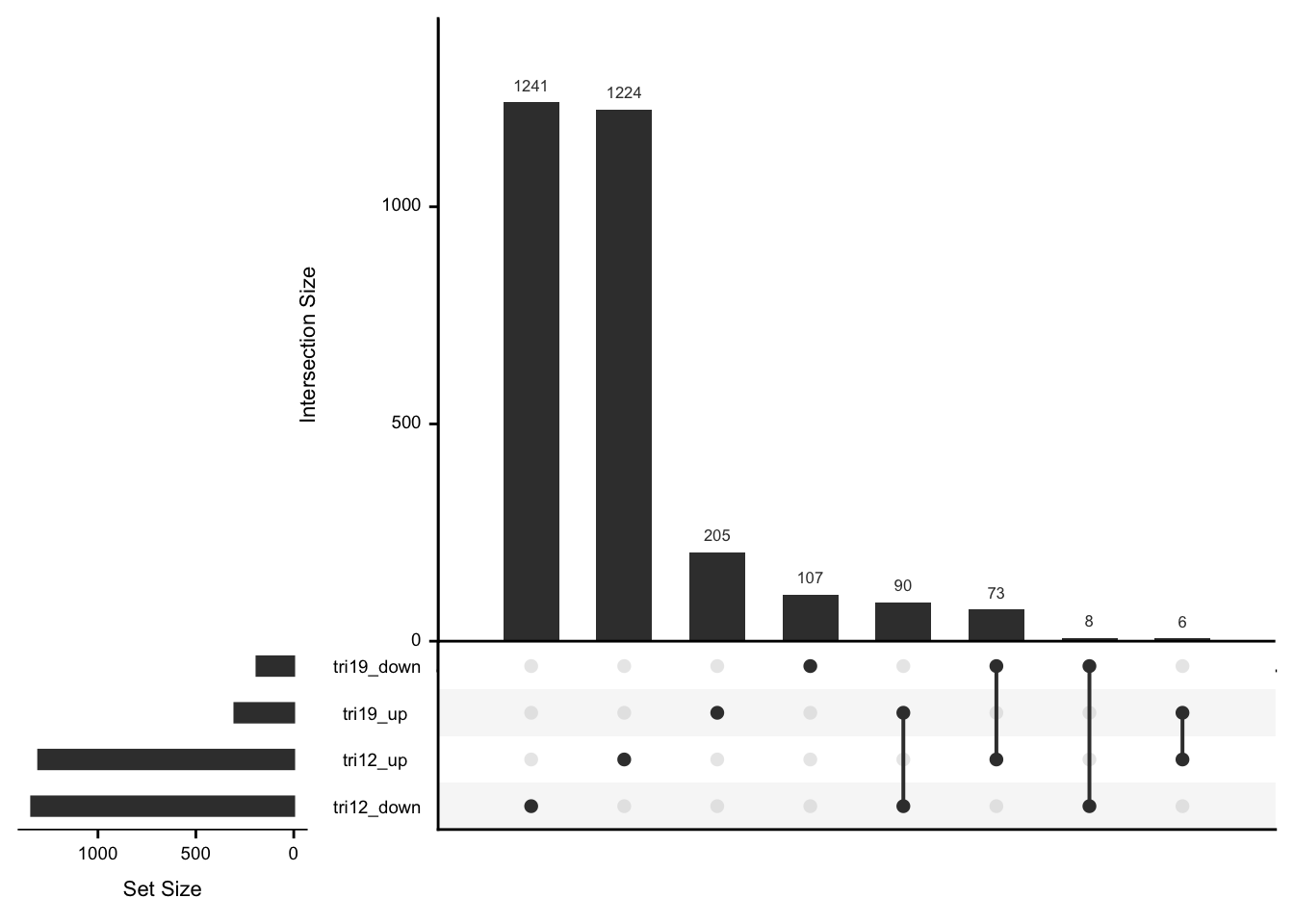
List of genes that up-regluated in trisomy12 and further up-regulated by the additional presence of trisomy19
compareTab <- full_join(resTab.tri12.sig, resTab.tri19.sig, by =c("name","id")) %>%
select(name, id, logFC.x, P.Value.x, adj.P.Val.x, logFC.y, P.Value.y, adj.P.Val.y) %>%
dplyr::rename(logFC.tri12 = logFC.x, logFC.tri19 = logFC.y,
P.Value.tri12 = P.Value.x, P.Value.tri19 = P.Value.y,
adj.P.Val.tri12 = adj.P.Val.x, adj.P.Val.tri19 = adj.P.Val.y) %>%
mutate(chr = rowData(ddsSub[id,])$chromosome)showList <- filter(compareTab, logFC.tri12 > 0 & logFC.tri19 >0) %>%
mutate_if(is.numeric, formatC, digits=2)
DT::datatable(showList)plotList <- lapply(seq(nrow(showList)), function(i) {
id <- showList[i,]$id
plotTab <- tibble(patID = ddsSub.vst$PatID,
count = assay(ddsSub.vst)[id,],
group = factor(ddsSub.vst$group,levels = c("none","onlyTri12","both")))
chrLoc <- showList[i,]$chr
ggplot(plotTab, aes(x=group, y = count)) + geom_boxplot(aes(fill = group)) + geom_point() +
ggtitle(sprintf("%s (on chr%s)",showList[i,]$name, chrLoc)) +
ggrepel::geom_text_repel(data = filter(plotTab, group %in% c("onlyTri12","both")),aes(label = patID))+
theme(legend.position ="none")
})
plot_grid(plotlist= plotList, ncol =2)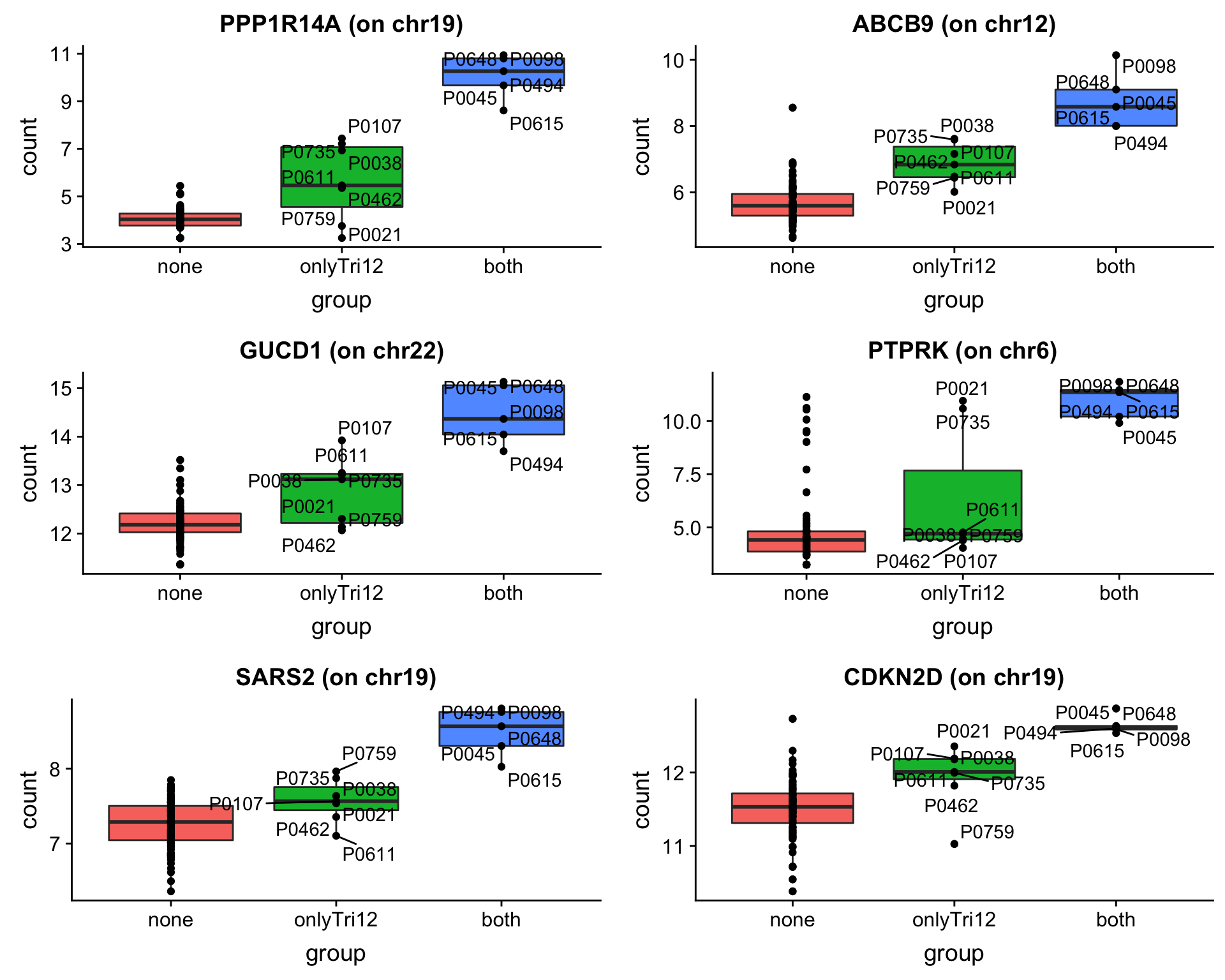
List of genes that down-regluated in trisomy12 and further down-regulated by the additional presence of trisomy19
showList <- filter(compareTab, logFC.tri12 < 0 & logFC.tri19 <0) %>%
mutate_if(is.numeric, formatC, digits=2)
DT::datatable(showList)plotList <- lapply(seq(nrow(showList)), function(i) {
id <- showList[i,]$id
plotTab <- tibble(patID = ddsSub.vst$PatID,
count = assay(ddsSub.vst)[id,],
group = factor(ddsSub.vst$group,levels = c("none","onlyTri12","both")))
chrLoc <- showList[i,]$chr
ggplot(plotTab, aes(x=group, y = count)) + geom_boxplot(aes(fill = group)) + geom_point() +
ggtitle(sprintf("%s (on chr%s)",showList[i,]$name, chrLoc)) +
ggrepel::geom_text_repel(data = filter(plotTab, group %in% c("onlyTri12","both")),aes(label = patID))+
theme(legend.position ="none")
})
plot_grid(plotlist= plotList, ncol =2)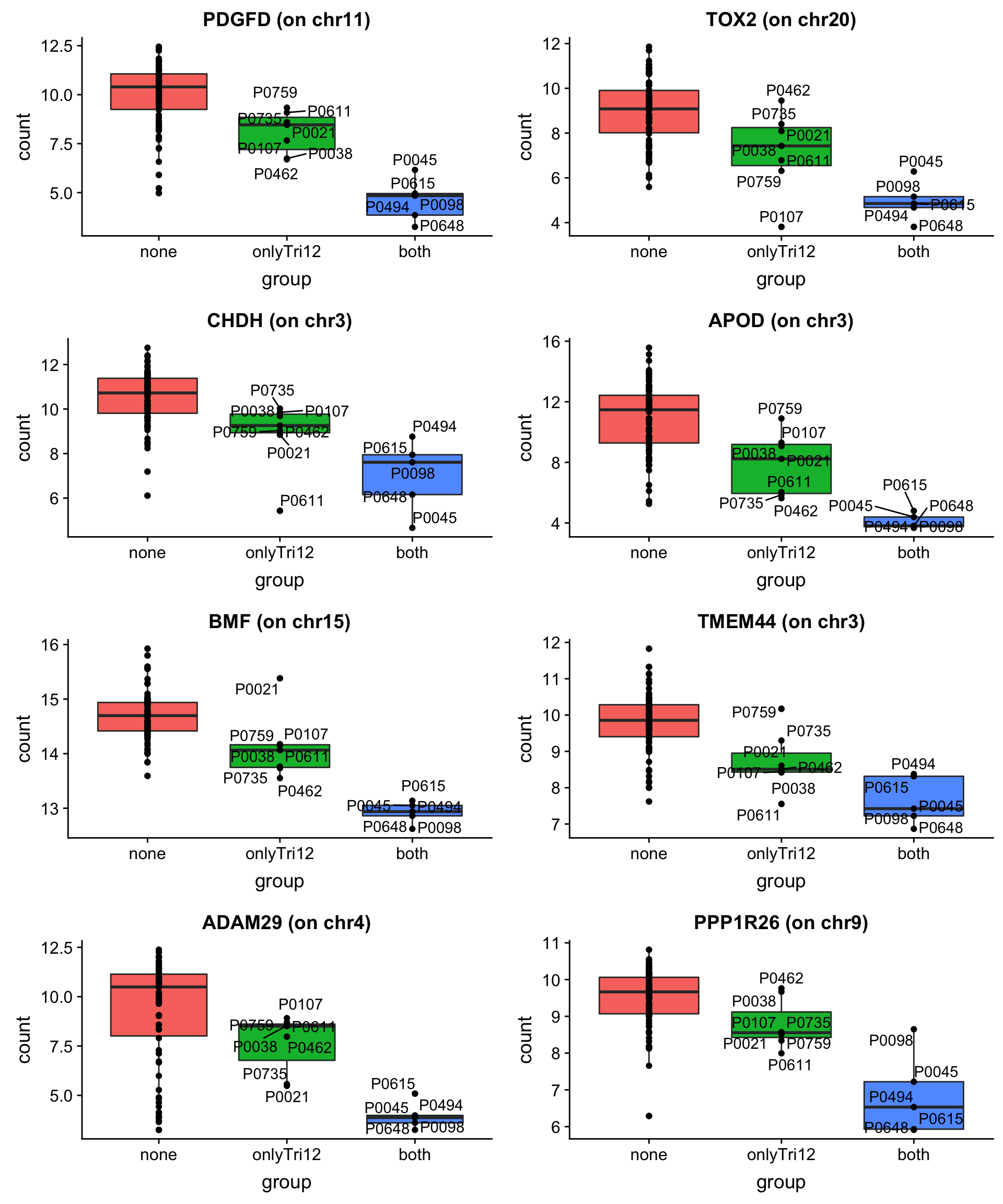
List of genes that up-regulated in trisomy12 and but down-regulated by the additional presence of trisomy19
showList <- filter(compareTab, logFC.tri12 > 0 & logFC.tri19 <0) %>%
arrange(desc(abs(logFC.tri19-logFC.tri12))) %>%
mutate_if(is.numeric, formatC, digits=2)
DT::datatable(showList)Example of top 9 (based on logFC differences) genes
plotList <- lapply(seq(9), function(i) {
id <- showList[i,]$id
plotTab <- tibble(patID = ddsSub.vst$PatID,
count = assay(ddsSub.vst)[id,],
group = factor(ddsSub.vst$group,levels = c("none","onlyTri12","both")))
chrLoc <- showList[i,]$chr
ggplot(plotTab, aes(x=group, y = count)) + geom_boxplot(aes(fill = group)) + geom_point() +
ggtitle(sprintf("%s (on chr%s)",showList[i,]$name, chrLoc)) +
ggrepel::geom_text_repel(data = filter(plotTab, group %in% c("onlyTri12","both")),aes(label = patID))+
theme(legend.position ="none")
})
plot_grid(plotlist= plotList, ncol =3)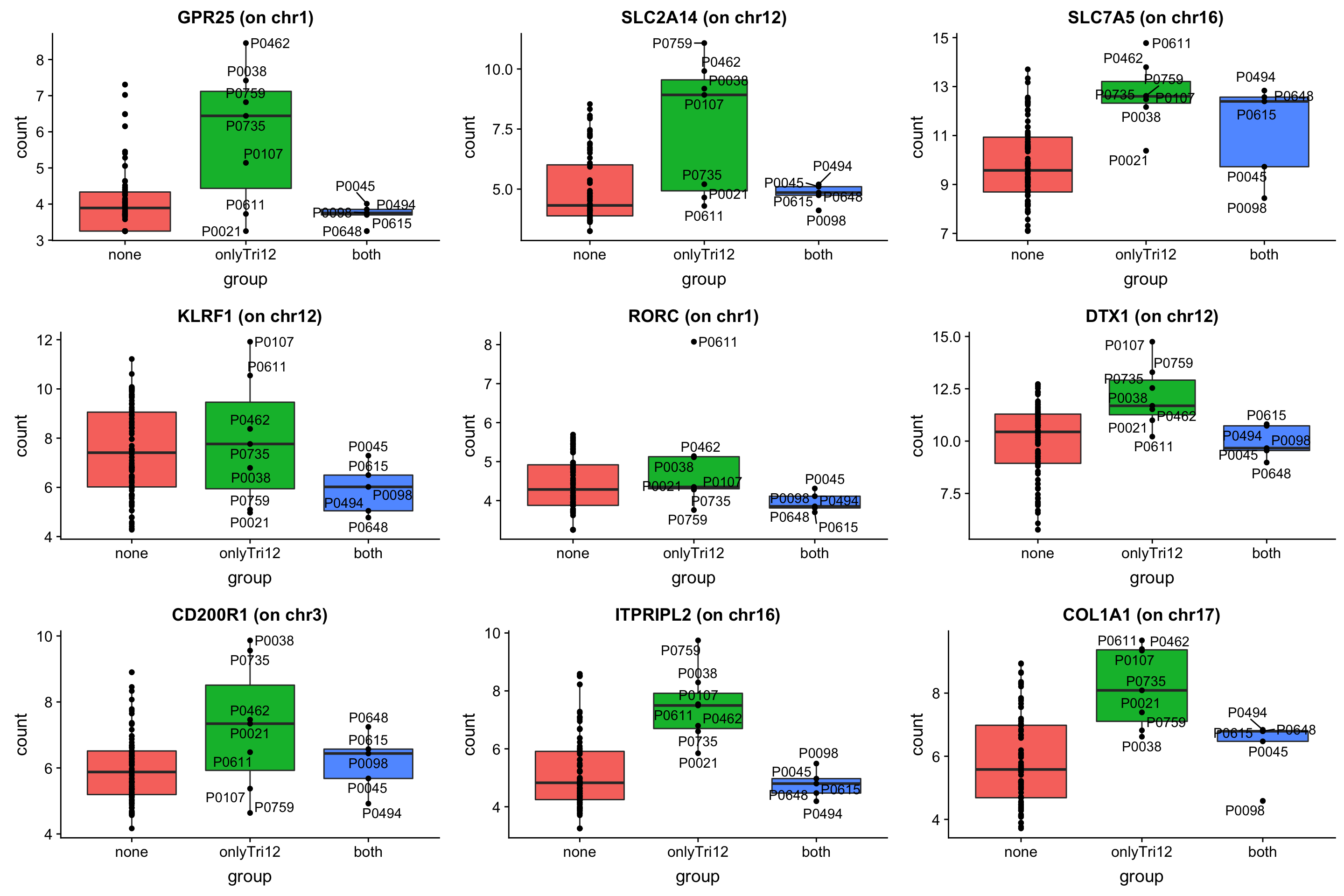
List of genes that down-regulated in trisomy12 and but up-regulated by the additional presence of trisomy19
showList <- filter(compareTab, logFC.tri12 < 0 & logFC.tri19 > 0) %>%
arrange(desc(abs(logFC.tri19-logFC.tri12))) %>%
mutate_if(is.numeric, formatC, digits=2)
DT::datatable(showList)Example of top 9 (based on logFC differences) genes
plotList <- lapply(seq(9), function(i) {
id <- showList[i,]$id
plotTab <- tibble(patID = ddsSub.vst$PatID,
count = assay(ddsSub.vst)[id,],
group = factor(ddsSub.vst$group,levels = c("none","onlyTri12","both")))
chrLoc <- showList[i,]$chr
ggplot(plotTab, aes(x=group, y = count)) + geom_boxplot(aes(fill = group)) + geom_point() +
ggtitle(sprintf("%s (on chr%s)",showList[i,]$name, chrLoc)) +
ggrepel::geom_text_repel(data = filter(plotTab, group %in% c("onlyTri12","both")),aes(label = patID))+
theme(legend.position ="none")
})
plot_grid(plotlist= plotList, ncol =3)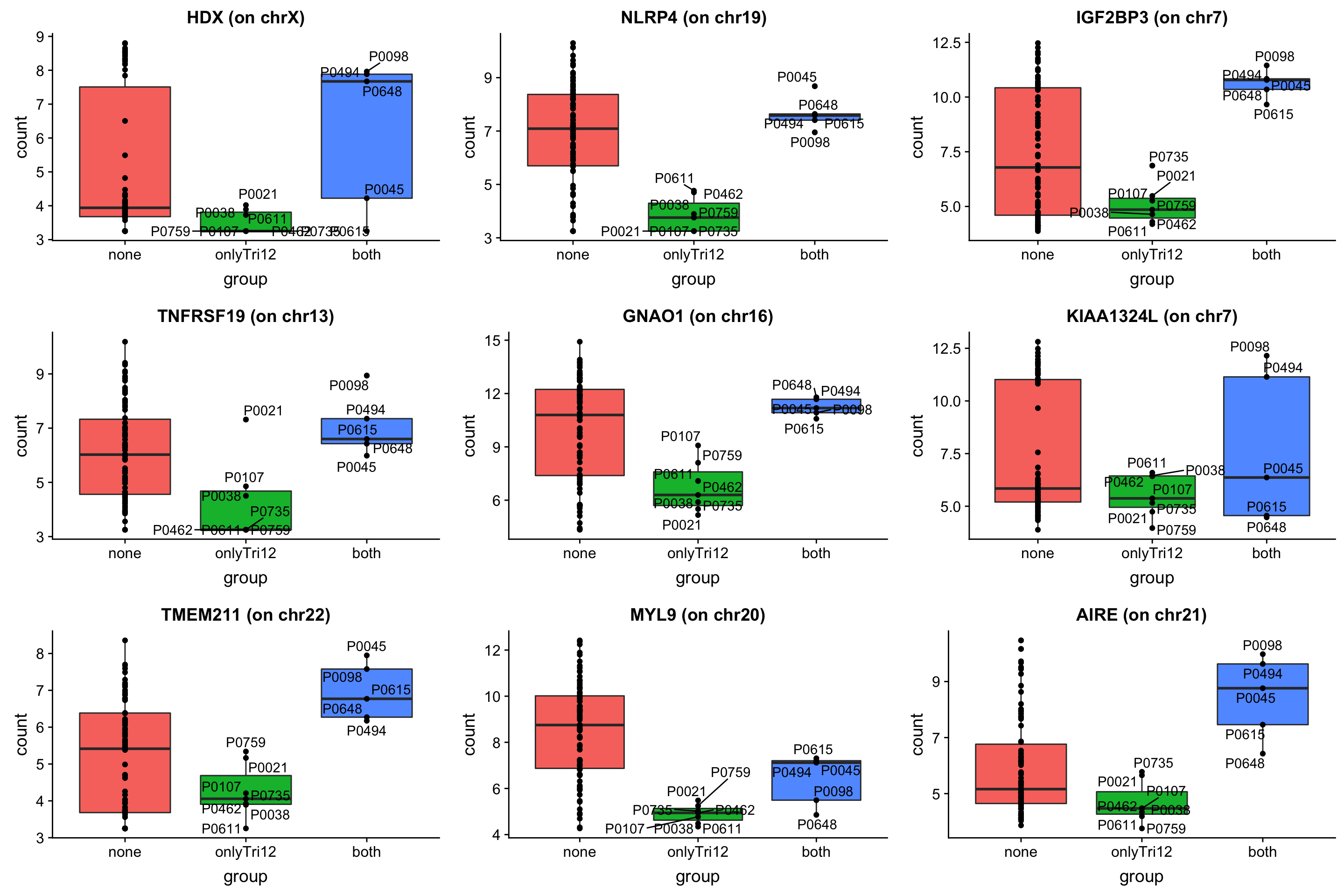
Analysis of differential drug responses related to trisomy19
Preprocessing
load("../../var/CPS1000_mainAnalysis.RData")
viabTab <- filter(pheno1000_main, !lowQuality, diagnosis %in% "CLL") %>%
left_join(select(patMeta, Patient.ID, IGHV.status, trisomy12, trisomy19), by = c(patientID = "Patient.ID")) %>%
filter(IGHV.status == "M", trisomy12 %in% 1, !is.na(trisomy19), !is.na(concIndex)) %>%
group_by(patientID, Drug, concIndex, Concentration, trisomy12, trisomy19) %>%
summarise(viab = mean(normVal.adj.sigm)) %>% ungroup() %>%
mutate(id = paste0(Drug,"_",concIndex))
screenData <- tidyToSum(viabTab, rowID = "id", colID = "patientID",
values = "viab",
annoRow = c("Drug","concIndex","Concentration"),
annoCol = c("trisomy12","trisomy19"))Sample distribution
annoTab <- tibble(patID = colnames(screenData),
tri12 = ifelse(screenData$trisomy12 == 1, "tri12","wt"),
tri19 = ifelse(screenData$trisomy19 ==1, "tri19","wt")) %>%
mutate(group = ifelse(tri12 == "tri12", ifelse(tri19 == "tri19","both","onlyTri12"),"none"))
table(annoTab$tri12, annoTab$tri19)
tri19 wt
tri12 5 7Add sample annotations
screenData$group <- annoTab[match(colnames(screenData),annoTab$patID),]$group
screenData <- screenData[,!is.na(screenData$group)]Differential drug responses related to trisomy19 in M-CLL samples with trisomy12
screenTest <- screenData[,!screenData$group %in% "none"]
designMat <- data.frame(colData(screenTest))
designMat <- model.matrix(~ trisomy19 ,designMat)T-test using Limma
testMat <- assay(screenTest)
fit <- lmFit(testMat, design = designMat)
fit2 <- eBayes(fit)
resTab.lm <- topTable(fit2, number ="all", adjust.method = "BH", coef = "trisomy191") %>%
data.frame() %>% rownames_to_column("id") %>%
mutate(Drug = rowData(screenData[id,])$Drug,
conc = rowData(screenData[id,])$Concentration)%>%
select(id, Drug, conc, logFC, t, P.Value, adj.P.Val) %>%
arrange(P.Value) %>%
as_tibble()P-value histogram
hist(resTab.lm$P.Value, breaks = 50)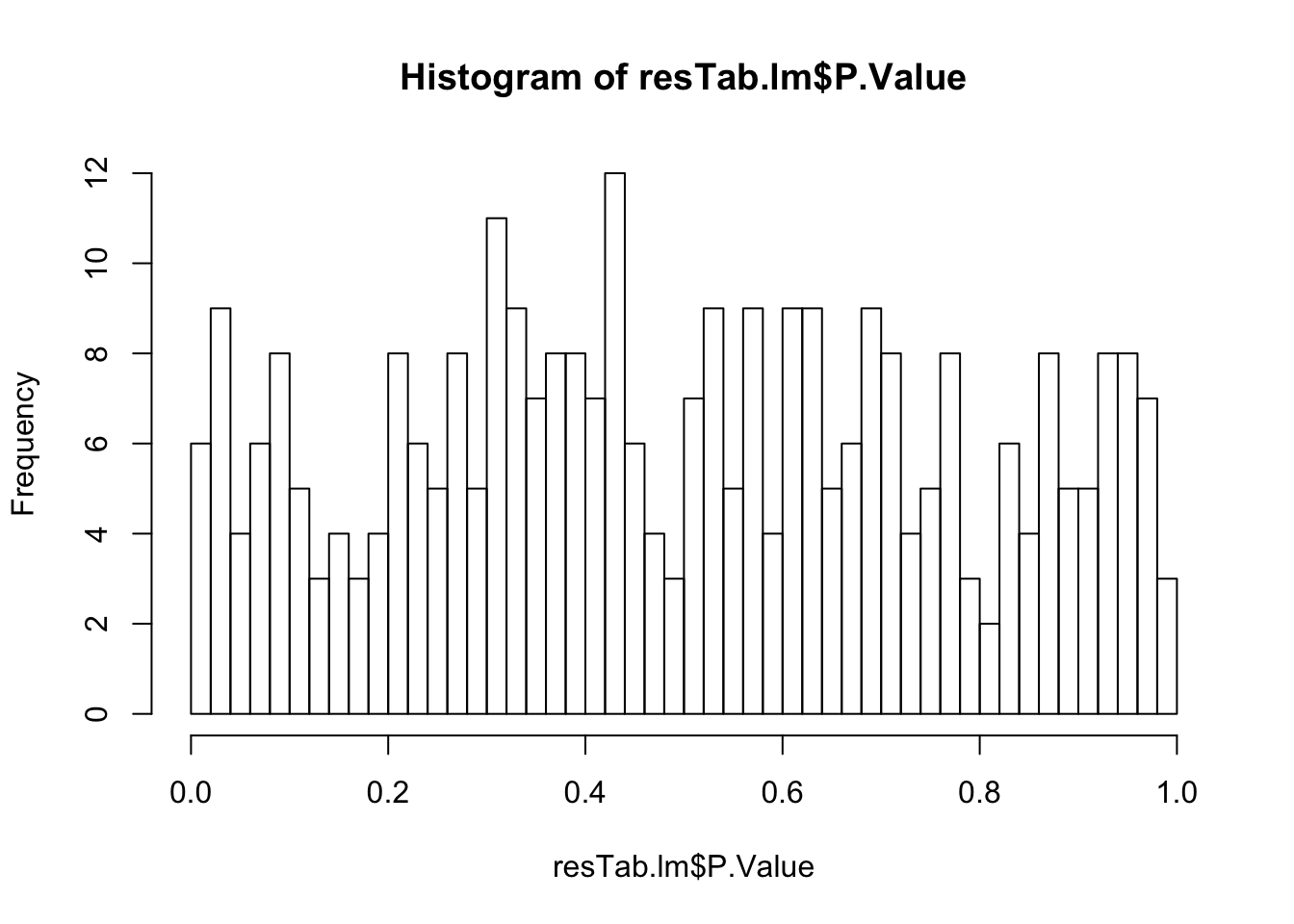 May not be many significant associations
May not be many significant associations
Table of associations with raw P-value < 0.05
corRes.sig <- resTab.lm %>% filter(P.Value <0.05)
corRes.sig %>% select(id, Drug, conc, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=2) %>% DT::datatable()Plot of significant assocations (raw P <0.05)
plotTab <- filter(viabTab, id %in% corRes.sig$id, trisomy12 %in% 1) %>%
mutate(id = factor(id, levels = corRes.sig$id))
ggplot(plotTab, aes(x=trisomy19, y = viab)) + geom_boxplot(aes(fill = trisomy19)) + geom_point() +
facet_wrap(~id, scale = "free", ncol=3) + ggrepel::geom_text_repel(aes(label = patientID))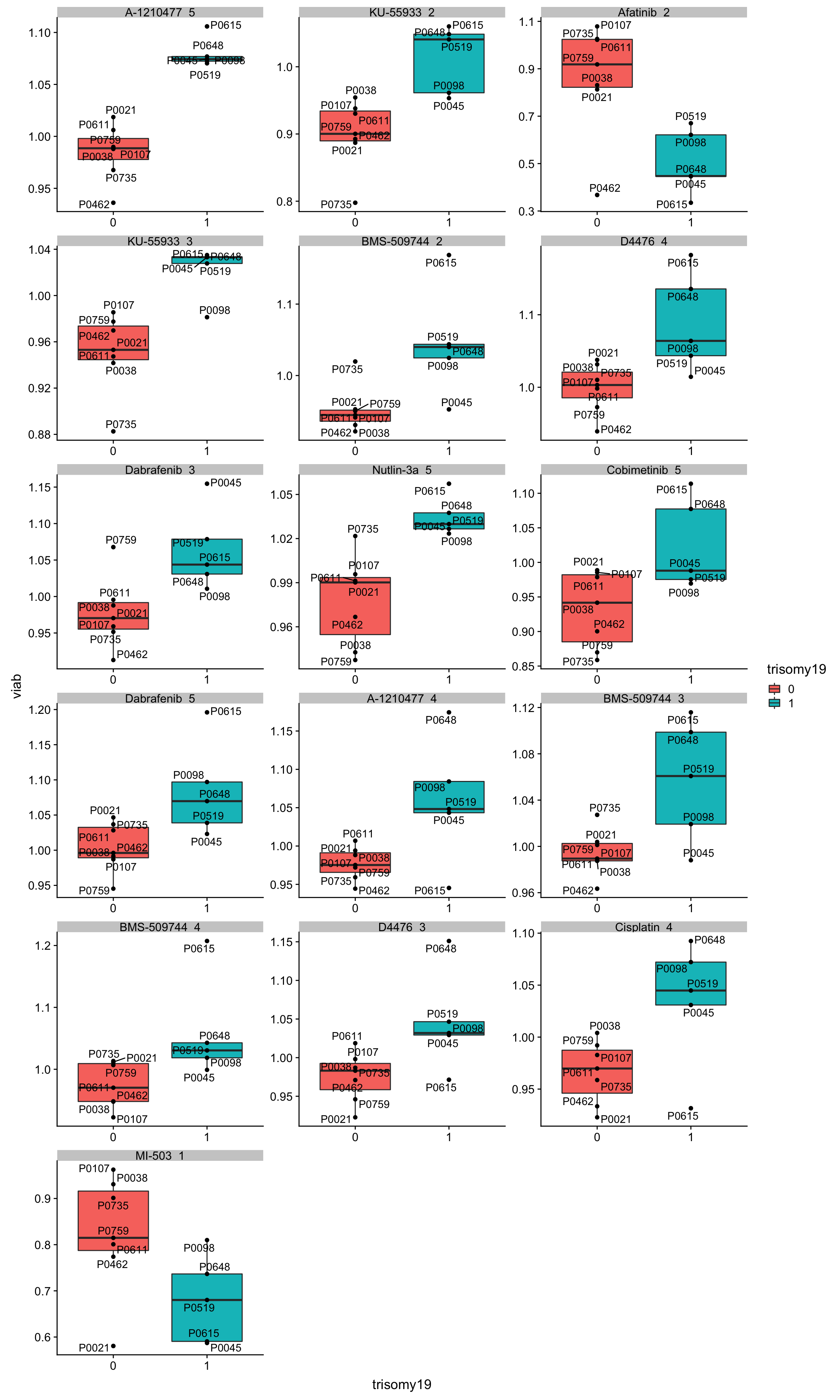 Although the individual P-Values are not very significant, but there’s a general trend that additional presence trisomy19 lead to drug resistance. But most of those associations happen at low concentrations and the overall effect size is not large. This maybe due to the baseline survival difference of samples with trisomy19.
Although the individual P-Values are not very significant, but there’s a general trend that additional presence trisomy19 lead to drug resistance. But most of those associations happen at low concentrations and the overall effect size is not large. This maybe due to the baseline survival difference of samples with trisomy19.
Compare baseline ATP value
load("../../var/basalATP_20190801.RData")
atpTab <- ATPcount.cps %>%
mutate(trisomy19 = patMeta[match(patientID, patMeta$Patient.ID),]$trisomy19) %>%
filter(IGHV == "M", trisomy12 == 1, !is.na(trisomy19)) %>%
select(patientID, ATPcount, day0, ATPdiff, ATPratio, trisomy19) %>%
mutate(log_ATP48h=log(ATPcount),log_ATPday0=log(day0),
ATPdiff=ATPcount - day0,
ATPratio = log_ATP48h - log_ATPday0) %>%
select(-ATPcount, -day0) %>%
gather(key = "measure", value = "val", -patientID, -trisomy19)
testRes <- group_by(atpTab, measure) %>%
nest() %>%
mutate(m = map(data, ~t.test(val ~ trisomy19,.,var.equal=TRUE))) %>%
mutate(res = map(m,broom::tidy)) %>%
unnest(res) %>%
select(measure, p.value)
testRes# A tibble: 4 x 2
# Groups: measure [4]
measure p.value
<chr> <dbl>
1 ATPdiff 0.0650
2 ATPratio 0.0513
3 log_ATP48h 0.0746
4 log_ATPday0 0.254 ggplot(atpTab, aes(x=trisomy19, y = val, fill = trisomy19)) + geom_boxplot() + geom_point() +
facet_wrap(~measure, scale="free") +
ggrepel::geom_text_repel(aes(label = patientID))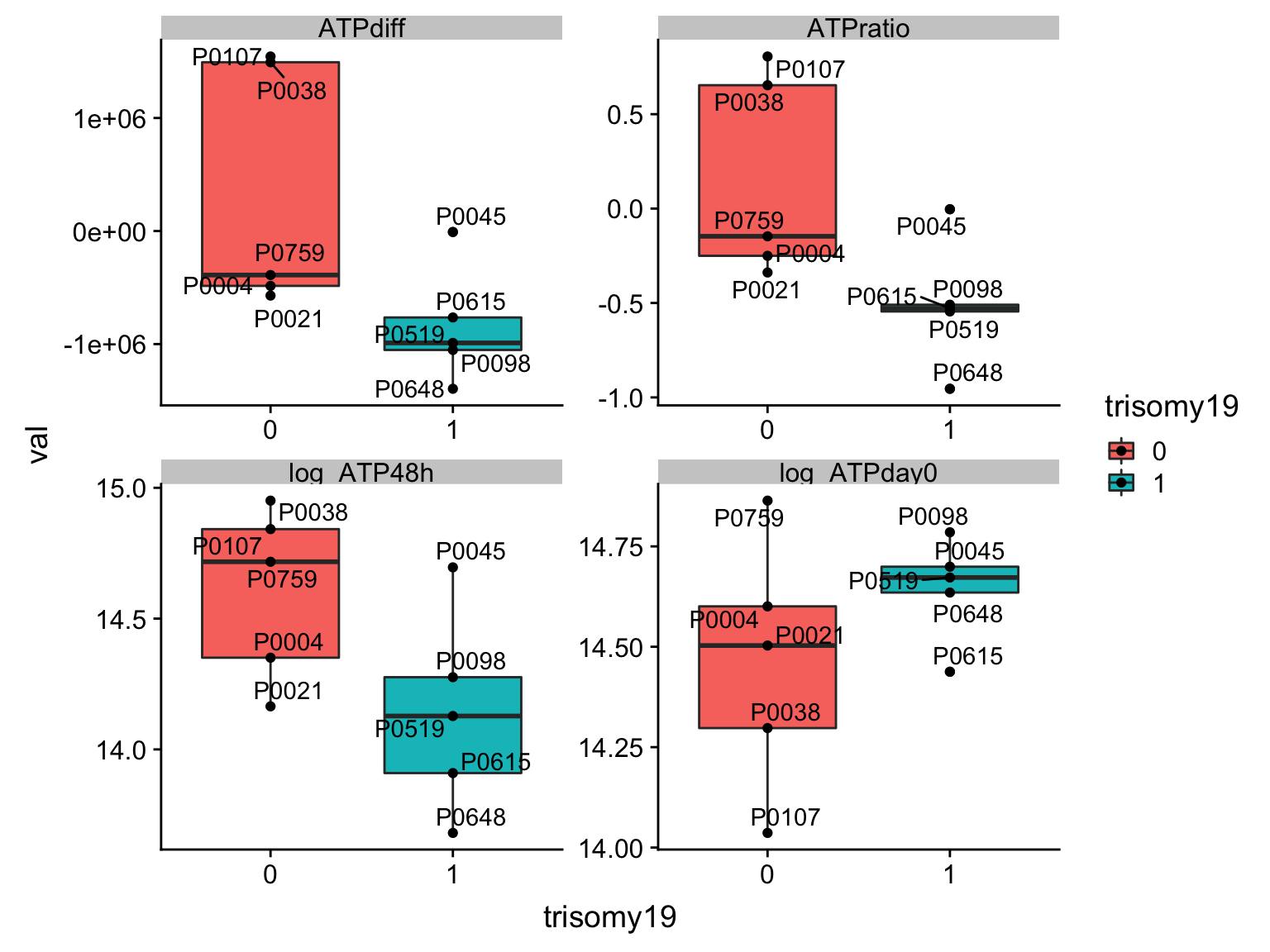 There’s a trend that samples with both trisomy19 and trisomy12 have higher day0 ATP, but tend to lose more viability during 48h culturing.
There’s a trend that samples with both trisomy19 and trisomy12 have higher day0 ATP, but tend to lose more viability during 48h culturing.
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] forcats_0.4.0 stringr_1.4.0
[3] dplyr_0.8.5 purrr_0.3.3
[5] readr_1.3.1 tidyr_1.0.0
[7] tibble_3.0.0 tidyverse_1.3.0
[9] jyluMisc_0.1.5 pheatmap_1.0.12
[11] UpSetR_1.4.0 limma_3.40.2
[13] DESeq2_1.24.0 SummarizedExperiment_1.14.0
[15] DelayedArray_0.10.0 BiocParallel_1.18.0
[17] matrixStats_0.54.0 Biobase_2.44.0
[19] GenomicRanges_1.36.0 GenomeInfoDb_1.20.0
[21] IRanges_2.18.1 S4Vectors_0.22.0
[23] BiocGenerics_0.30.0 piano_2.0.2
[25] proDA_1.1.2 cowplot_0.9.4
[27] ggplot2_3.3.0
loaded via a namespace (and not attached):
[1] utf8_1.1.4 shinydashboard_0.7.1 tidyselect_1.0.0
[4] RSQLite_2.1.1 AnnotationDbi_1.46.0 htmlwidgets_1.3
[7] grid_3.6.0 maxstat_0.7-25 munsell_0.5.0
[10] codetools_0.2-16 DT_0.7 withr_2.1.2
[13] colorspace_1.4-1 knitr_1.23 rstudioapi_0.10
[16] ggsignif_0.5.0 labeling_0.3 git2r_0.26.1
[19] slam_0.1-45 GenomeInfoDbData_1.2.1 KMsurv_0.1-5
[22] bit64_0.9-7 farver_2.0.3 rprojroot_1.3-2
[25] vctrs_0.2.4 generics_0.0.2 TH.data_1.0-10
[28] xfun_0.8 sets_1.0-18 R6_2.4.0
[31] locfit_1.5-9.1 bitops_1.0-6 fgsea_1.10.0
[34] assertthat_0.2.1 promises_1.0.1 scales_1.1.0
[37] multcomp_1.4-10 nnet_7.3-12 gtable_0.3.0
[40] extraDistr_1.8.11 sandwich_2.5-1 workflowr_1.6.0
[43] rlang_0.4.5 genefilter_1.66.0 cmprsk_2.2-8
[46] splines_3.6.0 acepack_1.4.1 broom_0.5.2
[49] checkmate_2.0.0 yaml_2.2.0 abind_1.4-5
[52] modelr_0.1.5 crosstalk_1.0.0 backports_1.1.4
[55] httpuv_1.5.1 Hmisc_4.2-0 tools_3.6.0
[58] relations_0.6-8 ellipsis_0.2.0 gplots_3.0.1.1
[61] RColorBrewer_1.1-2 Rcpp_1.0.1 plyr_1.8.4
[64] base64enc_0.1-3 visNetwork_2.0.7 zlibbioc_1.30.0
[67] RCurl_1.95-4.12 ggpubr_0.2.1 rpart_4.1-15
[70] zoo_1.8-6 haven_2.2.0 ggrepel_0.8.1
[73] cluster_2.1.0 exactRankTests_0.8-30 fs_1.4.0
[76] magrittr_1.5 data.table_1.12.2 openxlsx_4.1.0.1
[79] reprex_0.3.0 survminer_0.4.4 mvtnorm_1.0-11
[82] hms_0.5.2 shinyjs_1.0 mime_0.7
[85] evaluate_0.14 xtable_1.8-4 XML_3.98-1.20
[88] rio_0.5.16 readxl_1.3.1 gridExtra_2.3
[91] compiler_3.6.0 KernSmooth_2.23-15 crayon_1.3.4
[94] htmltools_0.4.0 later_0.8.0 Formula_1.2-3
[97] geneplotter_1.62.0 lubridate_1.7.4 DBI_1.0.0
[100] dbplyr_1.4.2 MASS_7.3-51.4 Matrix_1.2-17
[103] car_3.0-3 cli_1.1.0 marray_1.62.0
[106] gdata_2.18.0 igraph_1.2.4.1 pkgconfig_2.0.2
[109] km.ci_0.5-2 foreign_0.8-71 xml2_1.2.2
[112] annotate_1.62.0 XVector_0.24.0 drc_3.0-1
[115] rvest_0.3.5 digest_0.6.19 rmarkdown_1.13
[118] cellranger_1.1.0 fastmatch_1.1-0 survMisc_0.5.5
[121] htmlTable_1.13.1 curl_3.3 shiny_1.3.2
[124] gtools_3.8.1 lifecycle_0.2.0 nlme_3.1-140
[127] jsonlite_1.6 carData_3.0-2 fansi_0.4.0
[130] pillar_1.4.3 lattice_0.20-38 httr_1.4.1
[133] plotrix_3.7-6 survival_2.44-1.1 glue_1.3.2
[136] zip_2.0.2 bit_1.1-14 stringi_1.4.3
[139] blob_1.1.1 latticeExtra_0.6-28 caTools_1.17.1.2
[142] memoise_1.1.0