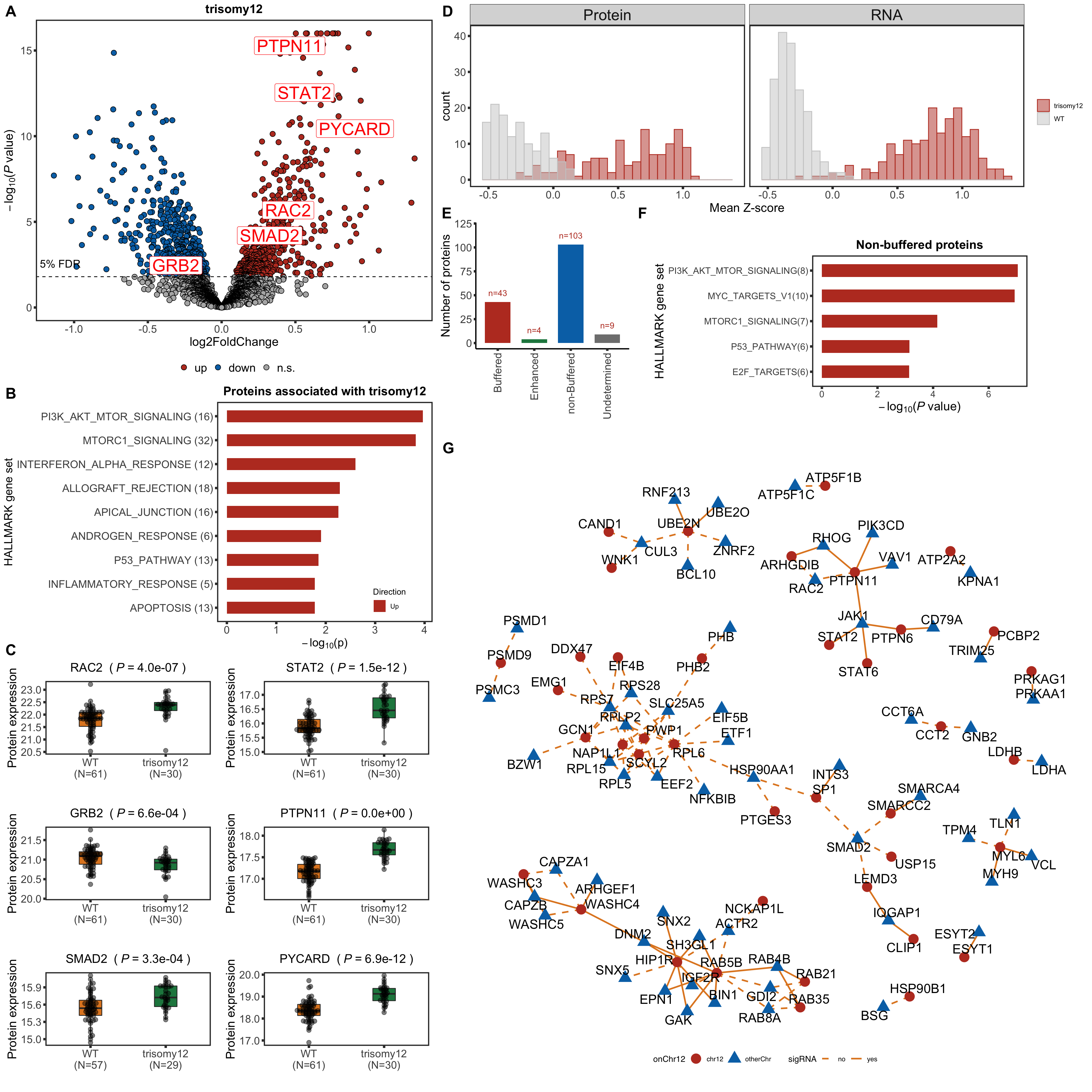
Visualizing gene dosage effect on protein and RNA level
Log2 protein counts
protExprTab <- sumToTidy(protCLL) %>%
filter(chromosome_name == "12") %>%
mutate(id = ensembl_gene_id, patID = colID, expr = log2Norm_combat, type = "Protein") %>%
select(id, patID, expr, type)
Log2 RNA seq counts
rnaExprTab <- counts(dds[rownames(dds) %in% protExprTab$id,
colnames(dds) %in% protExprTab$patID], normalized= TRUE) %>%
as_tibble(rownames = "id") %>%
pivot_longer(-id, names_to = "patID", values_to = "count") %>%
mutate(expr = log2(count)) %>%
select(id, patID, expr) %>% mutate(type = "RNA")
comExprTab <- bind_rows(rnaExprTab, protExprTab) %>%
mutate(trisomy12 = patMeta[match(patID, patMeta$Patient.ID),]$trisomy12) %>%
filter(!is.na(trisomy12)) %>% mutate(cnv = ifelse(trisomy12 %in% 1, "trisomy12","WT"))
Proteins/RNAs on Chr12 have higher expressions in trisomy12 samples compared to other samples
plotTab <- comExprTab %>%
group_by(id,type) %>% mutate(zscore = (expr-mean(expr))/sd(expr)) %>%
group_by(id, cnv, type) %>% summarise(meanExpr = mean(zscore, na.rm=TRUE)) %>%
ungroup()
dosagePlot <- ggplot(plotTab, aes(x=meanExpr, fill = cnv, col=cnv)) +
geom_histogram(position = "identity", alpha=0.5, bins=30) + facet_wrap(~type, scale = "fixed") +
scale_fill_manual(values = c(WT = "grey80", trisomy12 = colList[1]), name = "") +
scale_color_manual(values = c(WT = "grey80", trisomy12 = colList[1]), name = "") +
#xlim(-1,1) +
theme_full + xlab("Mean Z-score") +
theme(strip.text = element_text(size =20))
dosagePlot

The variation of expression is higher in RNA than protein
For proteins/RNA on chr12
plotTab <- comExprTab %>%
group_by(id, type) %>% summarise(varExp = sd(expr, na.rm=TRUE)) %>%
ungroup()
ggplot(plotTab, aes(x=varExp, fill = type, col=type)) +
geom_histogram(position = "identity", alpha=0.5) +
scale_fill_manual(values = c(RNA = colList[4], Protein = colList[5]), name = "") +
scale_color_manual(values = c(RNA = colList[4], Protein = colList[5]), name = "") +
theme_full + xlab("Standard deviation of expression")

The overall scale of change is higher in RNA expression than protein expression
plotTab <- comExprTab%>%
group_by(id, type, cnv) %>% summarise(meanExp = mean(expr, na.rm=TRUE)) %>%
ungroup() %>% spread(key = cnv, value = meanExp) %>%
mutate(log2FC = trisomy12-WT)
ggplot(plotTab, aes(x=log2FC, fill = type, col=type)) +
geom_histogram(position = "identity", alpha=0.5, bins = 100) +
scale_fill_manual(values = c(RNA = colList[3], Protein = colList[4]), name = "") +
scale_color_manual(values = c(RNA = colList[3], Protein = colList[4]), name = "") +
#coord_cartesian(xlim=c(-0.25,0.25))+
geom_vline(xintercept = 0, col = colList[1], linetype = "dashed") +
theme_full + xlab("log2 Fold Change")

Analyzing buffering effect
Detect buffered and non-buffered proteins
Preprocessing protein and RNA data
#subset samples and genes
overSampe <- intersect(colnames(ddsCLL), colnames(protCLL))
overGene <- intersect(rownames(ddsCLL), rowData(protCLL)$ensembl_gene_id)
ddsSub <- ddsCLL[overGene, overSampe]
protSub <- protCLL[match(overGene, rowData(protCLL)$ensembl_gene_id),overSampe]
rowData(ddsSub)$uniprotID <- rownames(protSub)[match(rownames(ddsSub),rowData(protSub)$ensembl_gene_id)]
#vst
ddsSub.vst <- ddsSub
assay(ddsSub.vst) <- log2(counts(ddsSub, normalized=TRUE) +1)
#ddsSub.vst <- varianceStabilizingTransformation(ddsSub)
Differential expression on RNA level
#design(ddsSub) <- ~ trisomy12 + IGHV
#deRes <- DESeq(ddsSub, betaPrior = TRUE)
rnaRes <- resListRNA %>% filter(Gene == "trisomy12") %>%
mutate(Chr = rowData(dds[id,])$chromosome) %>%
#filter(Chr == "12") %>%
#mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>%
dplyr::rename(geneID = id, log2FC.rna = log2FC,
pvalue.rna = P.Value, padj.rna = adj.P.Val, stat.rna= t) %>%
select(geneID, log2FC.rna, pvalue.rna, padj.rna, stat.rna)
Protein abundance changes related to trisomy12
protRes <- resList %>% filter(Gene == "trisomy12") %>%
mutate(Chr = rowData(protCLL[id,])$chromosome_name) %>%
#filter(Chr == "12") %>%
#mutate(adj.P.Val = p.adjust(P.Value, method= "BH")) %>%
dplyr::rename(uniprotID = id,
pvalue = P.Value, padj = adj.P.global,
chrom = Chr) %>%
mutate(geneID = rowData(protCLL[uniprotID,])$ensembl_gene_id) %>%
select(name, uniprotID, geneID, chrom, log2FC, pvalue, padj, t) %>%
dplyr::rename(stat =t) %>%
arrange(pvalue) %>% as_tibble()
Combine
allRes <- left_join(protRes, rnaRes, by = "geneID") %>%
filter(!is.na(stat), !is.na(stat.rna))
Only chr12 genes that are up-regulated are considered.
fdrCut <- 0.05
bufferTab <- allRes %>% filter(chrom %in% 12, stat.rna > 0, stat>0) %>%
ungroup() %>%
mutate(stat.prot.sqrt = sqrt(stat),
stat.prot.center = stat.prot.sqrt - mean(stat.prot.sqrt,na.rm= TRUE)) %>%
mutate(score = -stat.prot.center*stat.rna,
diffFC = log2FC.rna-log2FC) %>%
mutate(ifBuffer = case_when(
padj < fdrCut & padj.rna < fdrCut & stat > 0 ~ "non-Buffered",
padj > fdrCut & padj.rna < fdrCut ~ "Buffered",
padj < fdrCut & padj.rna > fdrCut & stat > 0 ~ "Enhanced",
TRUE ~ "Undetermined"
)) %>%
arrange(desc(score))
Table
bufferTab %>% select(name, geneID, chrom, ifBuffer, score, log2FC, padj, log2FC.rna, padj.rna) %>%
mutate_if(is.numeric, formatC, digits=2) %>%
DT::datatable()
Summary plot
sumTab <- bufferTab %>% group_by(ifBuffer) %>%
summarise(n = length(name))
bufferPlot <- ggplot(sumTab, aes(x=ifBuffer, y = n)) +
geom_bar(aes(fill = ifBuffer), stat="identity", width = 0.7) +
geom_text(aes(label = paste0("n=", n)),vjust=-1,col=colList[1]) +
scale_fill_manual(values =c(Buffered = colList[1],
Enhanced = colList[4],
`non-Buffered` = colList[2],
Undetermined = "grey50")) +
theme_half + theme(axis.text.x = element_text(angle = 90, hjust=1, vjust=0.5),
legend.position = "none") +
ylab("Number of proteins") + ylim(0,120) +xlab("")
bufferPlot

Plot example cases of buffered and non-buffered proteins
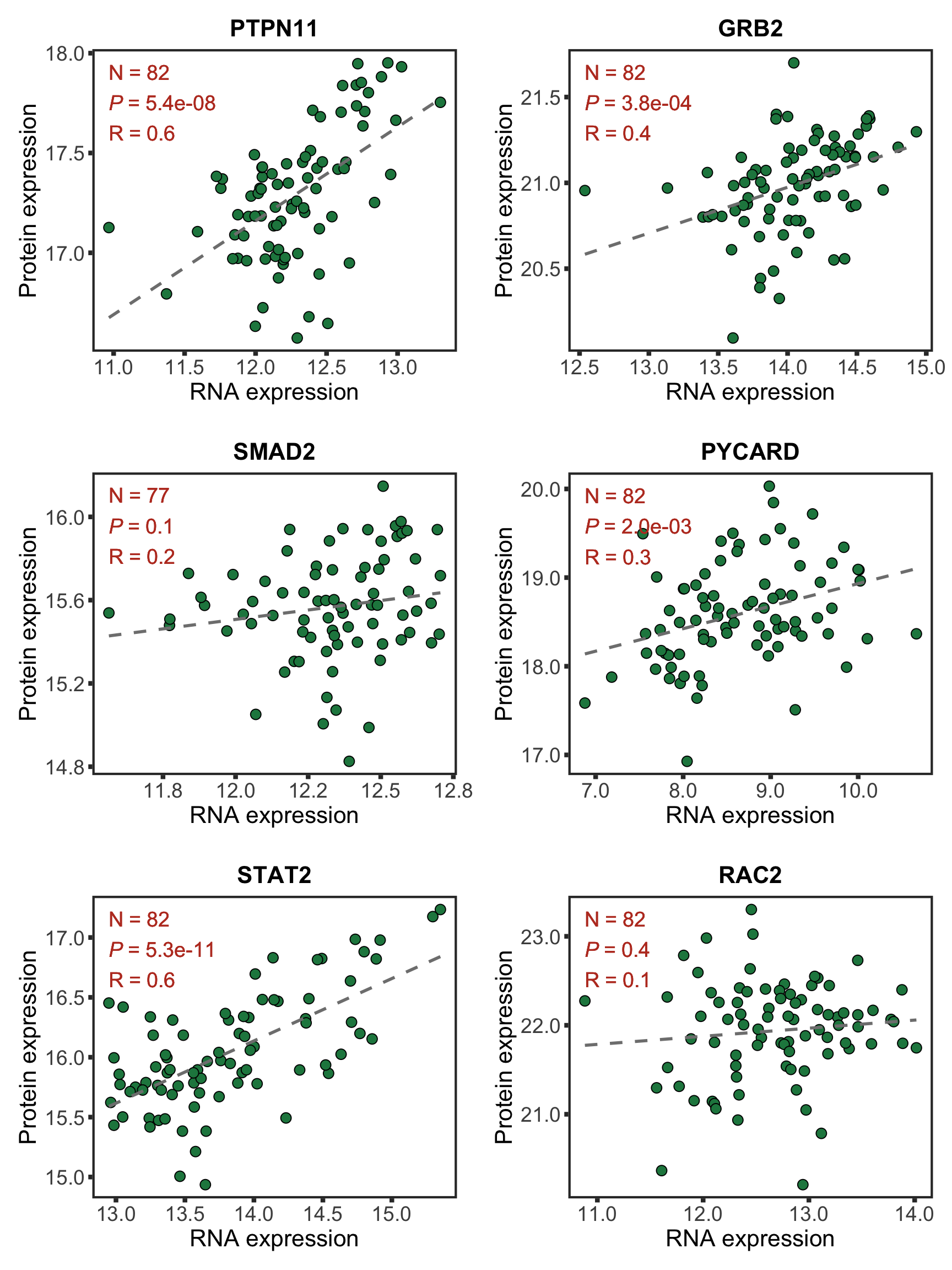
protList <- c("PTPN11","STAT2","CD27","SUDS3")
geneList <- bufferTab[match(protList, bufferTab$name),]$geneID
pList <- lapply(geneList, function(i) {
tabProt <- protExprTab %>% filter(id == i) %>%
select(id, patID,expr) %>% dplyr::rename(protExpr = expr)
tabRna <- rnaExprTab %>% filter(id == i) %>%
select(id, patID, expr) %>% dplyr::rename(rnaExpr = expr)
plotTab <- left_join(tabProt, tabRna, by = c("id","patID")) %>%
filter(!is.na(protExpr), !is.na(rnaExpr)) %>%
mutate(trisomy12 = patMeta[match(patID, patMeta$Patient.ID),]$trisomy12) %>%
mutate(trisomy12 = ifelse(trisomy12 %in% 1, "yes","no")) %>%
mutate(symbol = rowData(dds[id,])$symbol)
p <- ggplot(plotTab, aes(x=rnaExpr, y = protExpr)) +
geom_point(aes(col=trisomy12)) +
geom_smooth(formula = y~x, method="lm",se=FALSE, color = "grey50", linetype ="dashed" ) +
ggtitle(unique(plotTab$symbol)) +
ylab("Protein expression") + xlab("RNA expression") +
scale_color_manual(values =c(yes = colList[1],no=colList[2])) +
theme_full + theme(legend.position = "bottom")
ggExtra::ggMarginal(p, type = "histogram", groupFill = TRUE)
})
cowplot::plot_grid(plotlist = pList, ncol=2)

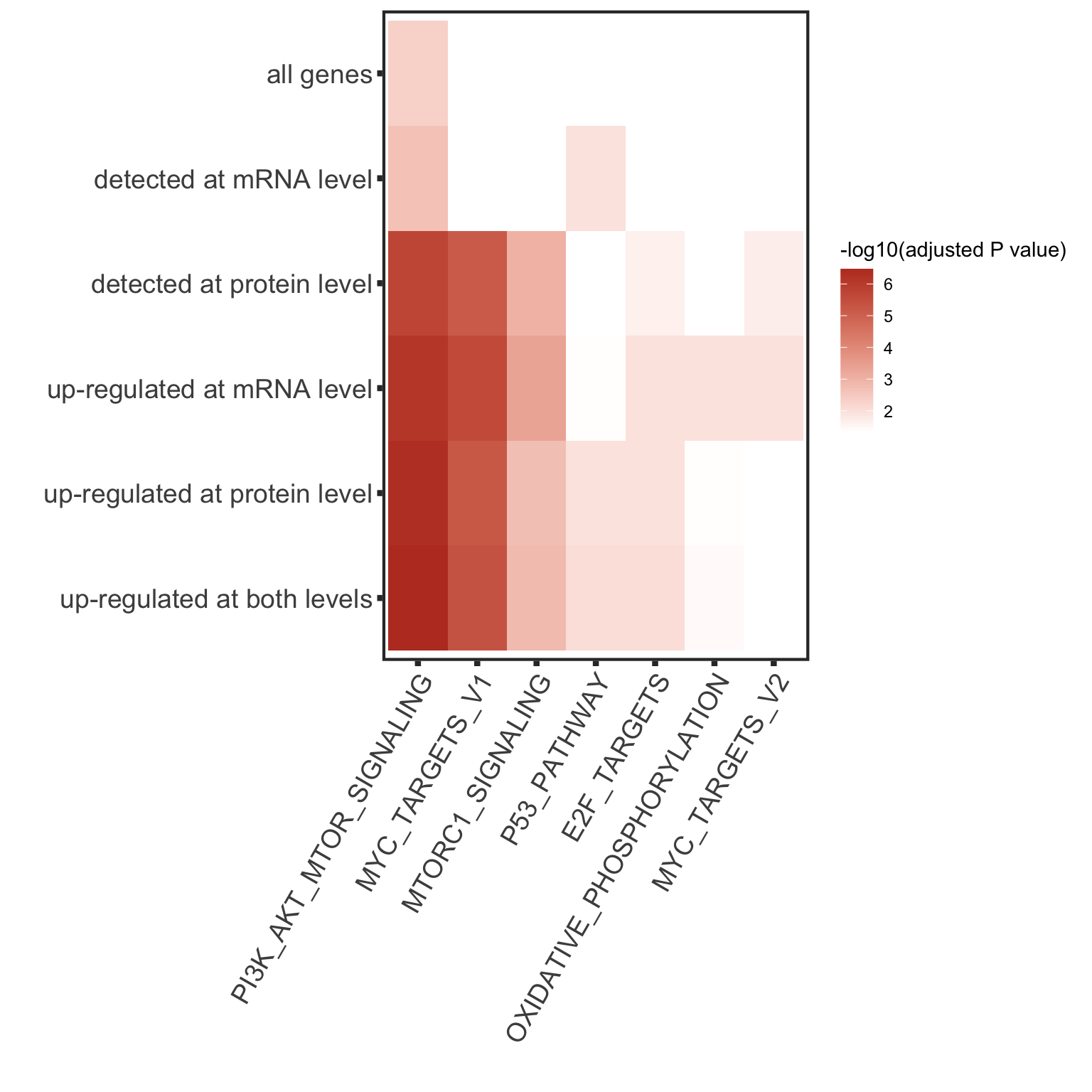
Enrichment of buffer and non-buffered proteins
Non-buffered prpteins
Using cancer hallmark genesets
rnaAll <- dds[rowData(dds)$biotype %in% "protein_coding" & !rowData(dds)$symbol %in% c("",NA),] #all protein coding gene as background
protList <- filter(bufferTab, ifBuffer == "non-Buffered")$name
refList <- rowData(rnaAll)$symbol
enRes <- runFisher(protList, refList, gmts$H, pCut =0.01, ifFDR = TRUE,removePrefix = "HALLMARK_",
plotTitle = "Non-buffered proteins", insideLegend = TRUE,
setName = "HALLMARK gene set")
bufferEnrich <- enRes$enrichPlot + theme(plot.margin = margin(1,3,1,1, unit = "cm"))
bufferEnrich

Buffered proteins
protList <- filter(bufferTab, ifBuffer == "Buffered")$name
enRes <- runFisher(protList, refList, gmts$H, pCut =0.1, ifFDR = TRUE)
[1] "No sets passed the criteria"
No enrichment
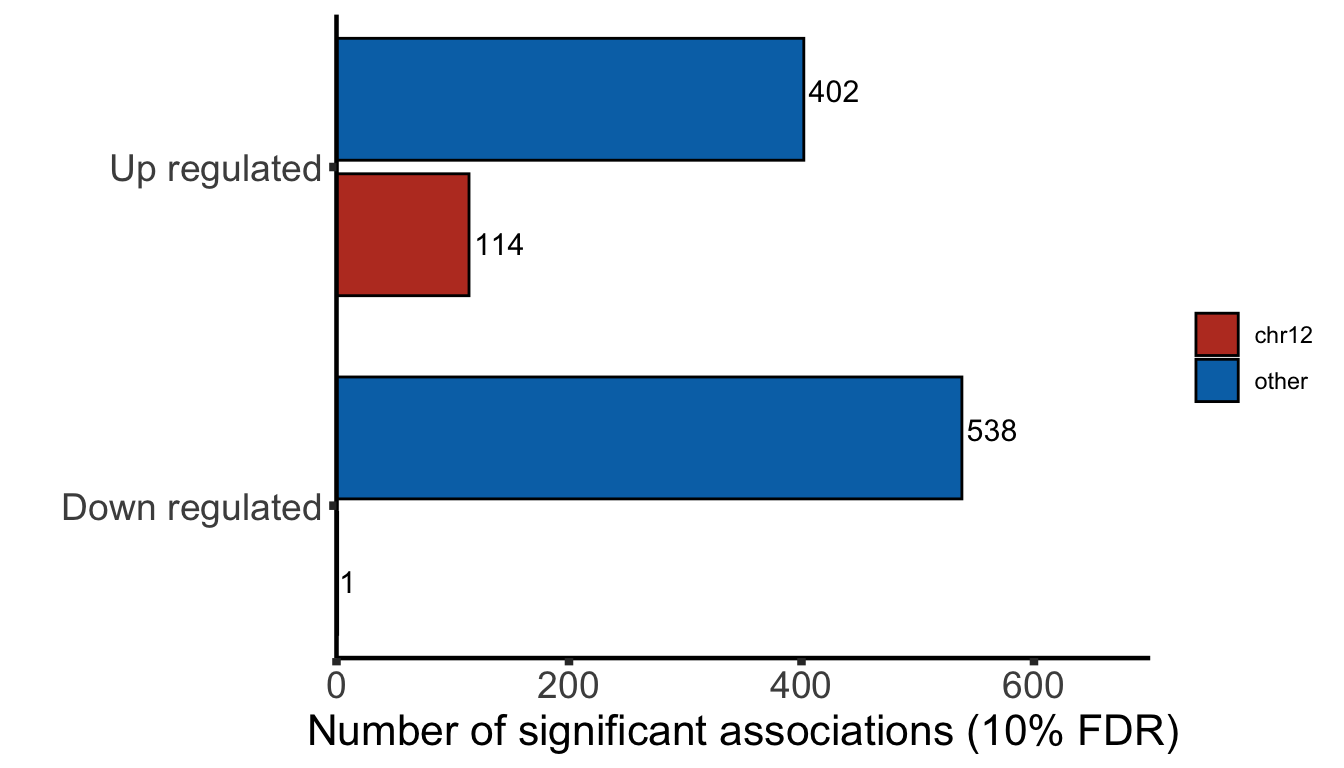
Compare buffering effect between trisomy19 and trisomy12
load("../output/deResList.RData")
load("../output/deResListRNA.RData")
testTabProt <- resList %>% mutate(chr = rowData(protCLL[id,])$chromosome_name) %>%
filter(Gene == paste0("trisomy",chr)) %>%
select(name, log2FC, Gene) %>% mutate(type = "Protein")
testTabRNA <- resListRNA %>% mutate(chr = rowData(dds[id,])$chromosome) %>%
filter(Gene == paste0("trisomy",chr)) %>%
select(name, log2FC, Gene) %>% mutate(type = "RNA")
overGene <- intersect(testTabProt$name, testTabRNA$name)
testTab <- bind_rows(testTabProt, testTabRNA) %>%
filter(name %in% overGene)
plotTab <- lapply(seq(-2,2, length.out = 50), function(foldCut) {
filTab <- mutate(testTab, pass = log2FC > foldCut) %>%
group_by(Gene, type) %>% summarise(n = sum(pass),per = sum(pass)/length(pass)) %>%
mutate(cut = foldCut)
}) %>% bind_rows() %>%
mutate(group =paste0(Gene,"_",type))
Cummulative plot
ggplot(plotTab, aes(x=cut, y = per))+
geom_line(aes(col = Gene, linetype = type),size=1) +
scale_color_manual(values = c(trisomy12 = colList[1],trisomy19=colList[2]), name = "") +
scale_linetype_discrete(name = "") +
coord_cartesian(xlim=c(1.5,-1)) +
ylab("Cumulative fraction") +
xlab("log2 (fold change)") +
theme_full +
theme(legend.position = c(0.8,0.3))

KS test
RNA level
testTab <- plotTab %>% filter(type == "RNA") %>%
select(Gene, per,cut) %>% pivot_wider(names_from = Gene, values_from = per)
ks.test(testTab$trisomy12, testTab$trisomy19)
Two-sample Kolmogorov-Smirnov test
data: testTab$trisomy12 and testTab$trisomy19
D = 0.24, p-value = 0.1122
alternative hypothesis: two-sided
Protein level
testTab <- plotTab %>% filter(type == "Protein") %>%
select(Gene, per,cut) %>% pivot_wider(names_from = Gene, values_from = per)
ks.test(testTab$trisomy12, testTab$trisomy19)
Two-sample Kolmogorov-Smirnov test
data: testTab$trisomy12 and testTab$trisomy19
D = 0.42, p-value = 0.0002955
alternative hypothesis: two-sided
Plot expression on genomic coordiate
patBack <- dplyr::filter(patMeta, Patient.ID %in% unique(allProtTab$patID)) %>%
dplyr::select(Patient.ID, trisomy12) %>%
dplyr::rename(patID = Patient.ID) %>%
mutate_all(as.character) %>%
mutate_at(vars(-patID),str_replace, "1","yes") %>%
mutate_at(vars(-patID),str_replace, "0","no")
plotExprVar <- function(gene, chr, patBack, allBand, allLine, allProtTab, allRnaTab, protLine = NULL,
region = c(-Inf,Inf),ifTrend = FALSE, normalize = TRUE, maxVal =2, minVal=-2) {
#table for cyto band
bandTab <- filter(allBand, ChromID == chr, chromStart >= region[1], chromEnd <= region[2]) %>%
mutate(chromMid = chromMid)
#table for expression
plotProtTab <- filter(allProtTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
plotRnaTab <- filter(allRnaTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
#summarise group mean
plotProtTab <- plotProtTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
plotRnaTab <- plotRnaTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
if (!is.null(protLine)) {
bufferLineTab <- plotProtTab %>%
select(symbol, mid_position, meanExpr, group) %>%
filter(symbol %in% protLine) %>%
pivot_wider(names_from = group, values_from = meanExpr) %>%
mutate(lowVal = map2_dbl(yes, no, min),
highVal = map2_dbl(yes, no, max))
} else bufferLineTab <- NULL
xMax <- max(bandTab$chromEnd, na.rm = T)
#main plot for Protein
gPro <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal+0.5, ymax=maxVal-0.5,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal-0.5, hjust =1, angle = 90, size=2.5)
if (!is.null(protLine)) {
gPro <- gPro + geom_segment(data = bufferLineTab, aes(x=mid_position, xend = mid_position,
y=lowVal, yend = highVal), linetype = "dashed")
}
gPro <- gPro + geom_rect(data = plotProtTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression (normalized by length)") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
yes = "darkred", no = "darkgreen")) +
scale_color_manual(values = c(yes = "darkred",no = "darkgreen")) +
ggtitle(paste0("Protein expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gPro <- gPro + geom_smooth(data =filter(plotProtTab, expr >0),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.5,
size =0.2, alpha=0.5)
}
#main plot for RNA
gRna <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal, ymax=maxVal,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal, hjust =1, angle = 90, size=2.5) +
geom_rect(data = plotRnaTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression Z-score") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
yes = "darkred", no = "darkgreen")) +
scale_color_manual(values = c(yes = "darkred", no = "darkgreen")) +
ggtitle(paste0("RNA expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gRna <- gRna + geom_smooth(data =filter(plotRnaTab),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.2,
size =0.2, alpha=0.5)
}
#for legend
## if the patient has CNV data
lgTab <- tibble(x= seq(6),y=seq(6),
Expression = c(rep("yes",3), rep("no",3)))
lg <- ggplot(lgTab, aes(x=x,y=y)) +
geom_point(aes(fill = Expression), shape =22,size=3) +
scale_fill_manual(values = c(yes = "darkred", no = "darkgreen"), name = gene) +
theme(legend.position = "bottom")
lg <- get_legend(lg)
return(list(plotPro = gPro, plotRNA = gRna, legend = lg))
}
Normalize protein and RNA expression
normalized <- TRUE
#if perform normalization
if (normalized) {
#for protein
exprMat <- select(allProtTab,patID, id,expr) %>%
distinct(patID, id, .keep_all = TRUE) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allProtTab <- select(allProtTab, -expr) %>% left_join(normTab, by = c("patID","id"))
#for RNA
exprMat <- select(allRnaTab,patID, id,expr) %>%
distinct(patID, id, .keep_all = TRUE) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allRnaTab <- select(allRnaTab, -expr) %>% left_join(normTab, by = c("patID","id"))
}
g <- plotExprVar("trisomy12","chr12",patBack,allBand, allLine,
allProtTab, allRnaTab, ifTrend = TRUE)
plot_grid(g$plotRNA, g$plotPro, g$legend, ncol = 1, rel_heights = c(1,1,0.2))


 PLCG1 is not significant anymore
PLCG1 is not significant anymore














 ### PCA
### PCA