Preprocessing
Preprocessing protein and RNA data
#subset samples and genes
overSampe <- intersect(colnames(ddsCLL), colnames(protCLL))
overGene <- intersect(rownames(ddsCLL), rowData(protCLL)$ensembl_gene_id)
ddsSub <- ddsCLL[overGene, overSampe]
protSub <- protCLL[match(overGene, rowData(protCLL)$ensembl_gene_id),overSampe]
rowData(ddsSub)$uniprotID <- rownames(protSub)[match(rownames(ddsSub),rowData(protSub)$ensembl_gene_id)]
#vst
ddsSub.vst <- varianceStabilizingTransformation(ddsSub)
Processing protein complex data
int_pairs <- int_pairs <- read_csv2("../output/int_pairs.csv")
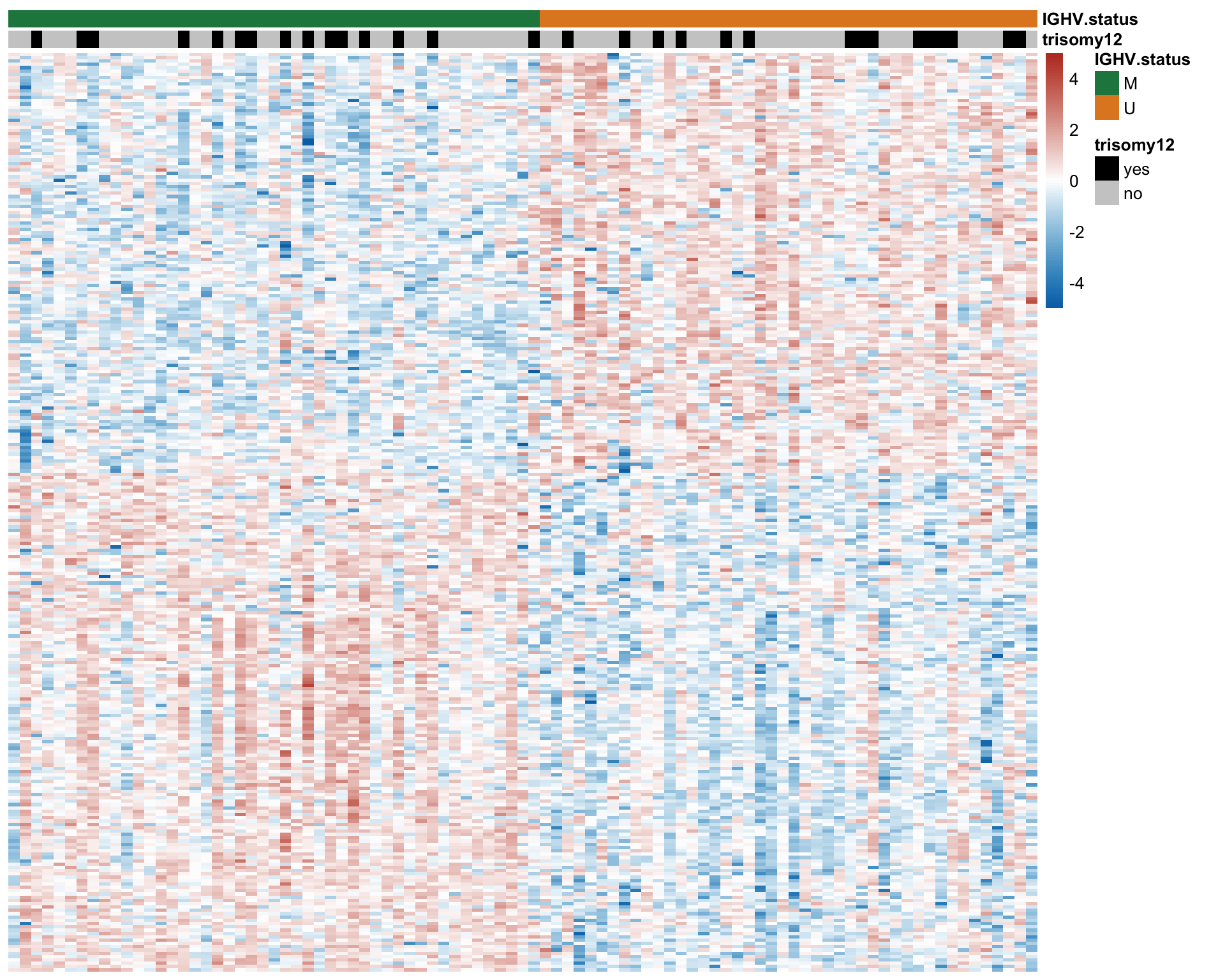
Differential RNA/protein expression analysis related to IGHV
Differential expression on RNA level
#design(ddsSub) <- ~ trisomy12 + IGHV
#deRes <- DESeq(ddsSub, betaPrior = TRUE)
rnaRes <- resListRNA %>% filter(Gene == "IGHV.status") %>%
mutate(Chr = rowData(dds[id,])$chromosome) %>%
#filter(Chr == "12") %>%
#mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>%
dplyr::rename(geneID = id, log2FC.rna = log2FC,
pvalue.rna = P.Value, padj.rna = adj.P.Val, stat.rna= t) %>%
select(geneID, log2FC.rna, pvalue.rna, padj.rna, stat.rna)
Number of differentially expressed RNA (10% FDR)
nrow(filter(rnaRes, padj.rna <0.05))
[1] 386
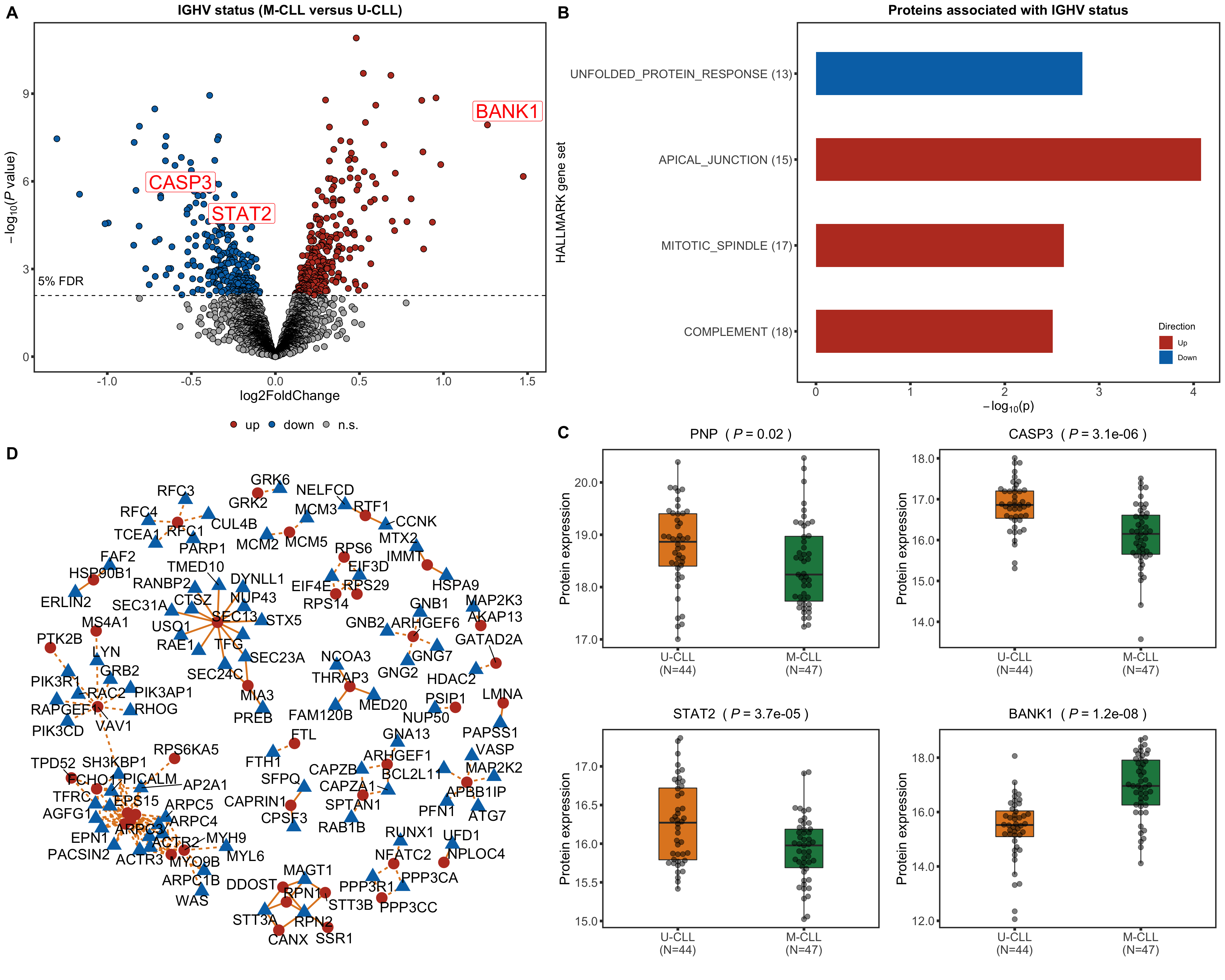
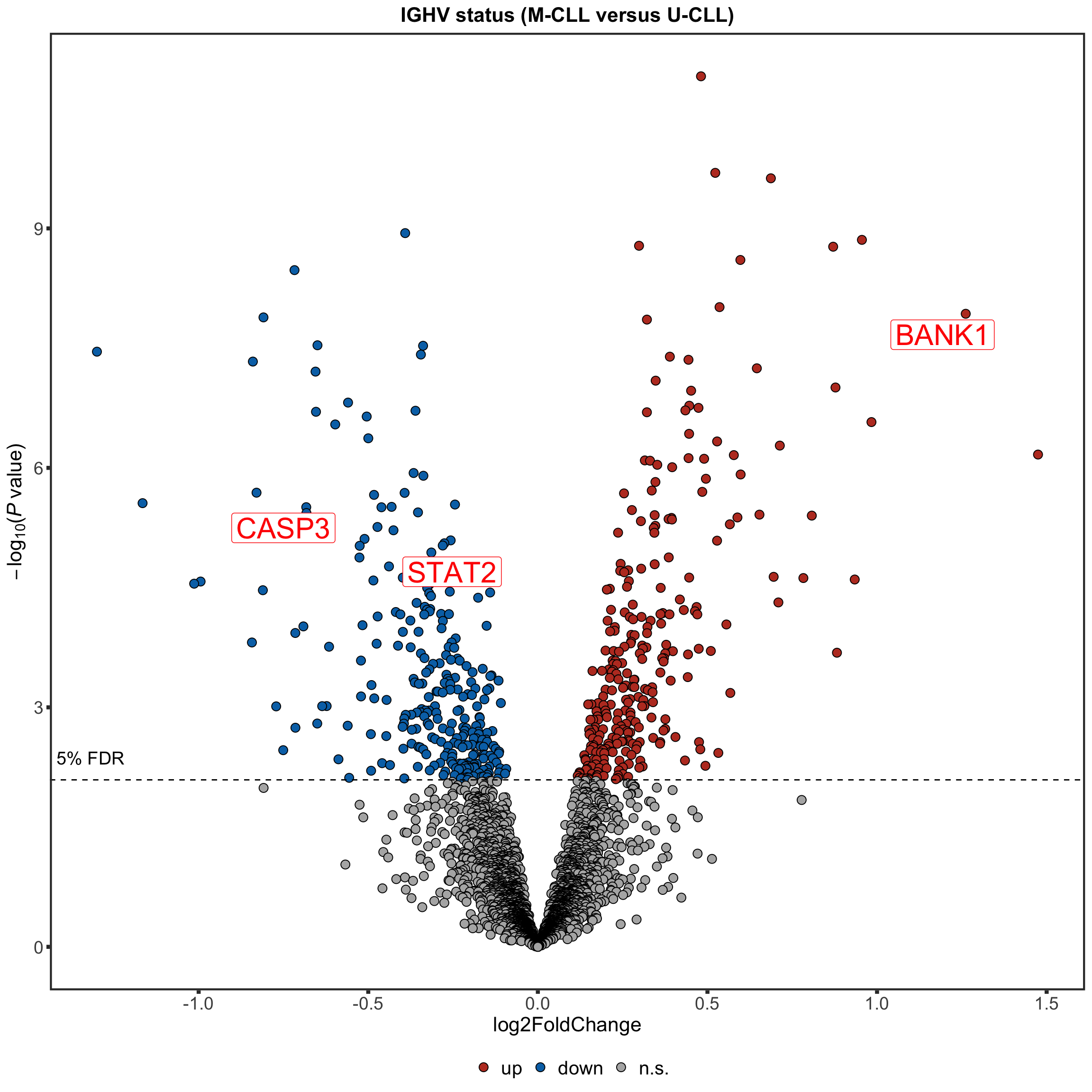
Protein abundance changes related to IGHV
fdrCut <- 0.05
protRes <- resList %>% filter(Gene == "IGHV.status") %>%
dplyr::rename(uniprotID = id,
pvalue = P.Value, padj = adj.P.Val,
chrom = Chr) %>%
mutate(geneID = rowData(protCLL[uniprotID,])$ensembl_gene_id) %>%
select(name, uniprotID, geneID, chrom, logFC, pvalue, padj) %>%
arrange(pvalue) %>% as_tibble()
Combine
allRes <- left_join(protRes, rnaRes, by = "geneID")
Construct protein-protein interaction network by “cause” proteins and “effect” proteins
fdrCut <- 0.05
resTab <- select(allRes, name, uniprotID, chrom, padj, padj.rna, logFC, log2FC.rna) %>%
mutate(sigProt = padj <= fdrCut,
sigRna = padj.rna <=fdrCut,
upProt = logFC > 0,
upRna = log2FC.rna >0)
comTab <- int_pairs %>% select(ProtA, ProtB, database) %>%
left_join(resTab, by = c(ProtA = "uniprotID")) %>%
left_join(resTab, by = c(ProtB = "uniprotID"))
comTab.filter <- comTab %>%
filter(sigProt.x, sigProt.y, logFC.x*logFC.y >0) %>%
mutate(direct = ifelse(logFC.x >0, "stabilizing", "destabilizing")) %>%
mutate(source = case_when(
sigProt.x & sigRna.x & sigProt.y & !sigRna.y ~ name.x,
sigProt.y & sigRna.y & sigProt.x & !sigRna.x ~ name.y)) %>%
filter(!is.na(source)) %>%
mutate(target = ifelse(name.x == source, name.y, name.x)) %>%
select(source, target, direct)
#get node list
allNodes <- union(comTab.filter$source, comTab.filter$target)
nodeList <- data.frame(id = seq(length(allNodes))-1, name = allNodes, stringsAsFactors = FALSE) %>%
mutate(causal = ifelse(name %in% comTab.filter$source, "cause", "effect"))
#get edge list
edgeList <- select(comTab.filter, source, target, direct) %>%
dplyr::rename(Source = source, Target = target) %>%
mutate(Source = nodeList[match(Source,nodeList$name),]$id,
Target = nodeList[match(Target, nodeList$name),]$id) %>%
data.frame(stringsAsFactors = FALSE)
net <- graph_from_data_frame(vertices = nodeList, d=edgeList, directed = FALSE)
tidyNet <- as_tbl_graph(net)
complexNet <- ggraph(tidyNet, layout = "igraph", algorithm = "nicely") +
geom_edge_link(aes(linetype = direct),color = colList[3], width=1) +
geom_node_point(aes(color =causal, shape = causal), size=6) +
geom_node_text(aes(label = name), repel = TRUE, size=6) +
scale_color_manual(values = c(cause = colList[1],effect = colList[2])) +
scale_linetype_manual(values = c(stabilizing = "solid", destabilizing = "dashed"))+
scale_edge_color_brewer(palette = "Set2") +
theme_graph(base_family = "sans") + theme(legend.position = "none")
complexNet

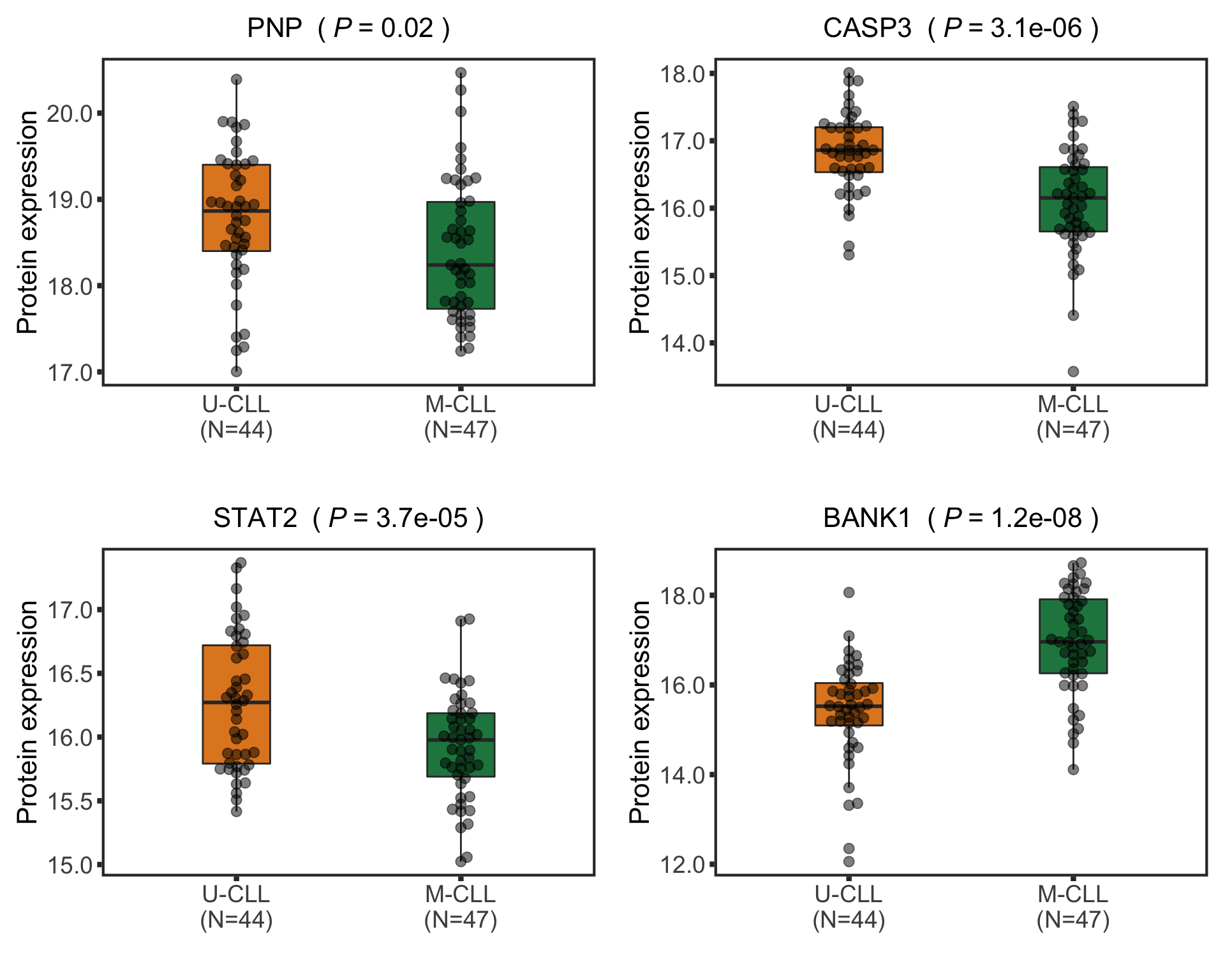
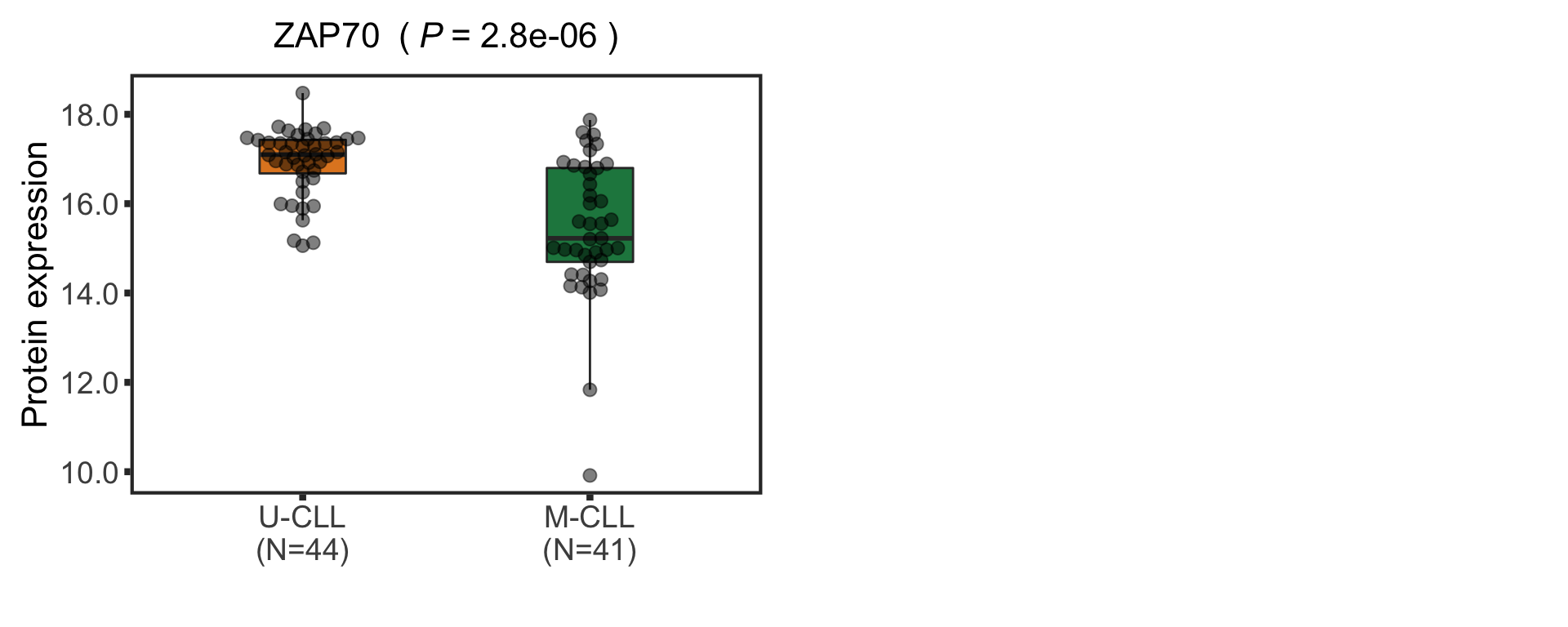 CD38 are not detected any more
CD38 are not detected any more