Feature selection with LASSO
Preprocessing data
Proteomics data
expVar <- "STAT2"
protMat <- assays(protCLL)[["QRILC_combat"]]
rownames(protMat) <- rowData(protCLL)$hgnc_symbol
yVec <- protMat[expVar,]
protMat <- protMat[rownames(protMat) != expVar,]
## Pre-filter for significant associations
designMat <- model.matrix(~yVec)
fit <- lmFit(protMat, design = designMat)
fit2 <- eBayes(fit)
resTab <- topTable(fit2, number = Inf)
keepProt <- filter(resTab, adj.P.Val < 0.1)$ID
protMat <- t(protMat[keepProt, ])
dim(protMat)
[1] 91 627
responseList <- list()
responseList[["STAT2"]] <- yVec
colnames(protMat) <- paste0(colnames(protMat),"_protein")
RNAseq
#subset
ddsSub <- dds[,dds$PatID %in% colnames(protCLL)]
#only keep protein coding genes with symbol
ddsSub <- ddsSub[rowData(ddsSub)$biotype %in% "protein_coding" & !rowData(ddsSub)$symbol %in% c("",NA),]
#remove lowly expressed genes
ddsSub <- ddsSub[rowSums(counts(ddsSub, normalized = TRUE)) > 100,]
#voom transformation
exprMat <- limma::voom(counts(ddsSub), lib.size = ddsSub$sizeFactor)$E
ddsSub.voom <- ddsSub
assay(ddsSub.voom) <- exprMat
rnaMat <- exprMat
rownames(rnaMat) <- rowData(ddsSub.voom)$symbol
# Prefiltering
overSampe <- intersect(names(yVec), colnames(rnaMat))
designMat <- model.matrix(~ yVec[overSampe])
fit <- lmFit(rnaMat[,overSampe], design = designMat)
fit2 <- eBayes(fit)
resTab <- topTable(fit2, number = Inf) %>% data.frame() %>% rownames_to_column("ID")
keepRna <- filter(resTab, adj.P.Val < 0.05)$ID
rnaMat <- t(rnaMat[keepRna, ])
dim(rnaMat)
[1] 82 704
colnames(rnaMat) <- paste0(colnames(rnaMat),"_rna")
Genomic data
ighvMap <- c(M = 1, U=0)
methMap <- c(LP= 0, IP=0.5, HP=1 )
#genetics
genData <- filter(patMeta, Patient.ID %in% colnames(protCLL)) %>%
select(-HIPO.ID, -project, -date.of.diagnosis, -treatment, -date.of.first.treatment,
-gender, -diagnosis, -Methylation_Cluster) %>%
dplyr::rename(IGHV = IGHV.status) %>%
mutate_at(vars(-Patient.ID), as.character) %>%
mutate(IGHV = ighvMap[IGHV]) %>%
mutate_at(vars(-Patient.ID), as.numeric) %>%
data.frame() %>% column_to_rownames("Patient.ID")
#remove gene with higher than 40% missing values
genData <- genData[,colSums(is.na(genData))/nrow(genData) <= 0.4]
#remove genes with less than 5 mutated cases
genData <- genData[,colSums(genData, na.rm = TRUE) >= 5]
#fill the missing value with majority
genData <- apply(genData, 2, function(x) {
xVec <- x
avgVal <- mean(x,na.rm= TRUE)
if (avgVal >= 0.5) {
xVec[is.na(xVec)] <- 1
} else xVec[is.na(xVec)] <- 0
xVec
})
Drug responses
#choose the first sample
viabMat <- arrange(pheno1000_main, screenDate) %>%
filter(diagnosis == "CLL", patientID %in% colnames(protCLL)) %>%
distinct(patientID, Drug, concIndex, .keep_all = TRUE) %>%
filter(! Drug %in% c("DMSO","PBS")) %>%
mutate(id = paste0(Drug,"_",concIndex)) %>%
select(patientID, id, normVal.adj.sigm) %>%
spread(key = patientID, value = normVal.adj.sigm) %>%
data.frame() %>% column_to_rownames("id") %>%
as.matrix() %>% t()
Feature selection with LASSO penalty
#Functions for running glm
runGlm <- function(X, y, method = "ridge", repeats=20, folds = 3, lambda = "lambda.1se") {
modelList <- list()
lambdaList <- c()
varExplain <- c()
coefMat <- matrix(NA, ncol(X), repeats)
rownames(coefMat) <- colnames(X)
if (method == "lasso"){
alpha = 1
} else if (method == "ridge") {
alpha = 0
}
for (i in seq(repeats)) {
if (ncol(X) > 2) {
res <- cv.glmnet(X,y, type.measure = "mse", family="gaussian",
nfolds = folds, alpha = alpha, standardize = FALSE)
lambdaList <- c(lambdaList, res[[lambda]])
modelList[[i]] <- res
coefModel <- coef(res, s = lambda)[-1] #remove intercept row
coefMat[,i] <- coefModel
#calculate variance explained
y.pred <- predict(res, s = lambda, newx = X)
varExp <- cor(as.vector(y),as.vector(y.pred))^2
varExplain[i] <- ifelse(is.na(varExp), 0, varExp)
} else {
fitlm<-lm(y~., data.frame(X))
varExp <- summary(fitlm)$r.squared
varExplain <- c(varExplain, varExp)
}
}
list(modelList = modelList, lambdaList = lambdaList, varExplain = varExplain, coefMat = coefMat)
}
#function for scaling predictors
dataScale <- function(x, censor = NULL, robust = FALSE) {
#function to scale different variables
if (length(unique(na.omit(x))) <=3){
#a binary variable, change to -0.5 and 0.5 for 1 and 2
x - 0.5
} else {
if (robust) {
#continuous variable, centered by median and divied by 2*mad
mScore <- (x-median(x,na.rm=TRUE))/mad(x,na.rm=TRUE)
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
} else {
mScore <- (x-mean(x,na.rm=TRUE))/(sd(x,na.rm=TRUE))
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
}
}
}
#function to generate response vector and explainatory variable for each seahorse measurement
generateData <- function(responseList, inclSet, onlyCombine = FALSE, censor = NULL, robust = FALSE) {
allResponse <- list()
allExplain <- list()
for (measure in names(responseList)) {
y <- responseList[[measure]]
y <- y[!is.na(y)]
#get overlapped samples for each dataset
overSample <- names(y)
for (eachSet in inclSet) {
overSample <- intersect(overSample,rownames(eachSet))
}
y <- dataScale(y[overSample], censor = censor, robust = robust)
expTab <- list()
if ("Gene" %in% names(inclSet)) {
geneTab <- inclSet$Gene[overSample,]
#at least 3 mutated sample
geneTab <- geneTab[, colSums(geneTab) >= 3]
vecName <- sprintf("genetic(%s)", ncol(geneTab))
expTab[[vecName]] <- apply(geneTab,2,dataScale)
}
if ("RNA" %in% names(inclSet)){
rnaMat <- inclSet$RNA[overSample, ]
colnames(rnaMat) <- paste0("con.",colnames(rnaMat), sep = "")
vecName <- sprintf("RNA(%s)", ncol(rnaMat))
expTab[[vecName]] <- apply(rnaMat,2,dataScale, censor = censor, robust = robust)
}
if ("Protein" %in% names(inclSet)){
protMat <- inclSet$Protein[overSample, ]
colnames(protMat) <- paste0("con.",colnames(protMat), sep = "")
vecName <- sprintf("Protein(%s)", ncol(protMat))
expTab[[vecName]] <- apply(protMat,2,dataScale, censor = censor, robust = robust)
}
if ("Drug" %in% names(inclSet)){
drugMat <- inclSet$Drug[overSample, ]
colnames(drugMat) <- paste0("con.",colnames(drugMat), sep = "")
vecName <- sprintf("Drug(%s)", ncol(drugMat))
expTab[[vecName]] <- apply(drugMat,2,dataScale, censor = censor, robust = robust)
}
comboTab <- c()
for (eachSet in names(expTab)){
comboTab <- cbind(comboTab, expTab[[eachSet]])
}
vecName <- sprintf("all(%s)", ncol(comboTab))
expTab[[vecName]] <- comboTab
allResponse[[measure]] <- y
allExplain[[measure]] <- expTab
}
if (onlyCombine) {
#only return combined results, for feature selection
allExplain <- lapply(allExplain, function(x) x[length(x)])
}
return(list(allResponse=allResponse, allExplain=allExplain))
}
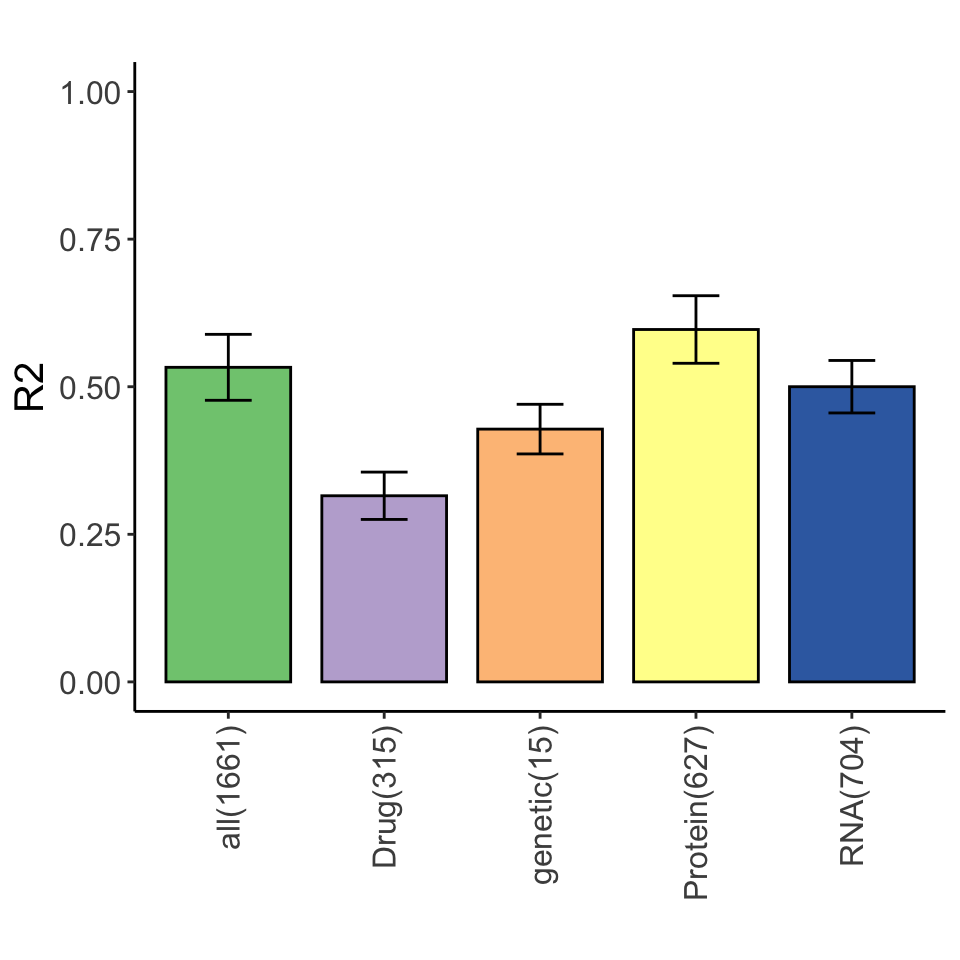
Clean and integrate multi-omics data
inclSet<-list(Gene=genData, RNA = rnaMat, Protein = protMat, Drug = viabMat)
cleanData <- generateData(responseList, inclSet, censor = 5)
#Function for multi-variate regression
runGlm <- function(X, y, method = "ridge", repeats=20, folds = 3) {
modelList <- list()
lambdaList <- c()
varExplain <- c()
coefMat <- matrix(NA, ncol(X), repeats)
rownames(coefMat) <- colnames(X)
if (method == "lasso"){
alpha = 1
} else if (method == "ridge") {
alpha = 0
}
for (i in seq(repeats)) {
if (ncol(X) > 2) {
res <- cv.glmnet(X,y, type.measure = "mse", family="gaussian",
nfolds = folds, alpha = alpha, standardize = FALSE)
lambdaList <- c(lambdaList, res$lambda.min)
modelList[[i]] <- res
coefModel <- coef(res, s = "lambda.min")[-1] #remove intercept row
coefMat[,i] <- coefModel
#calculate variance explained
y.pred <- predict(res, s = "lambda.min", newx = X)
varExp <- 1-min(res$cvm)/res$cvm[1]
#varExp <- cor(as.vector(y),as.vector(y.pred))^2
varExplain[i] <- ifelse(is.na(varExp), 0, varExp)
} else {
fitlm<-lm(y~., data.frame(X))
varExp <- summary(fitlm)$r.squared
varExplain <- c(varExplain, varExp)
}
}
list(modelList = modelList, lambdaList = lambdaList, varExplain = varExplain, coefMat = coefMat)
}
set.seed(2020)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
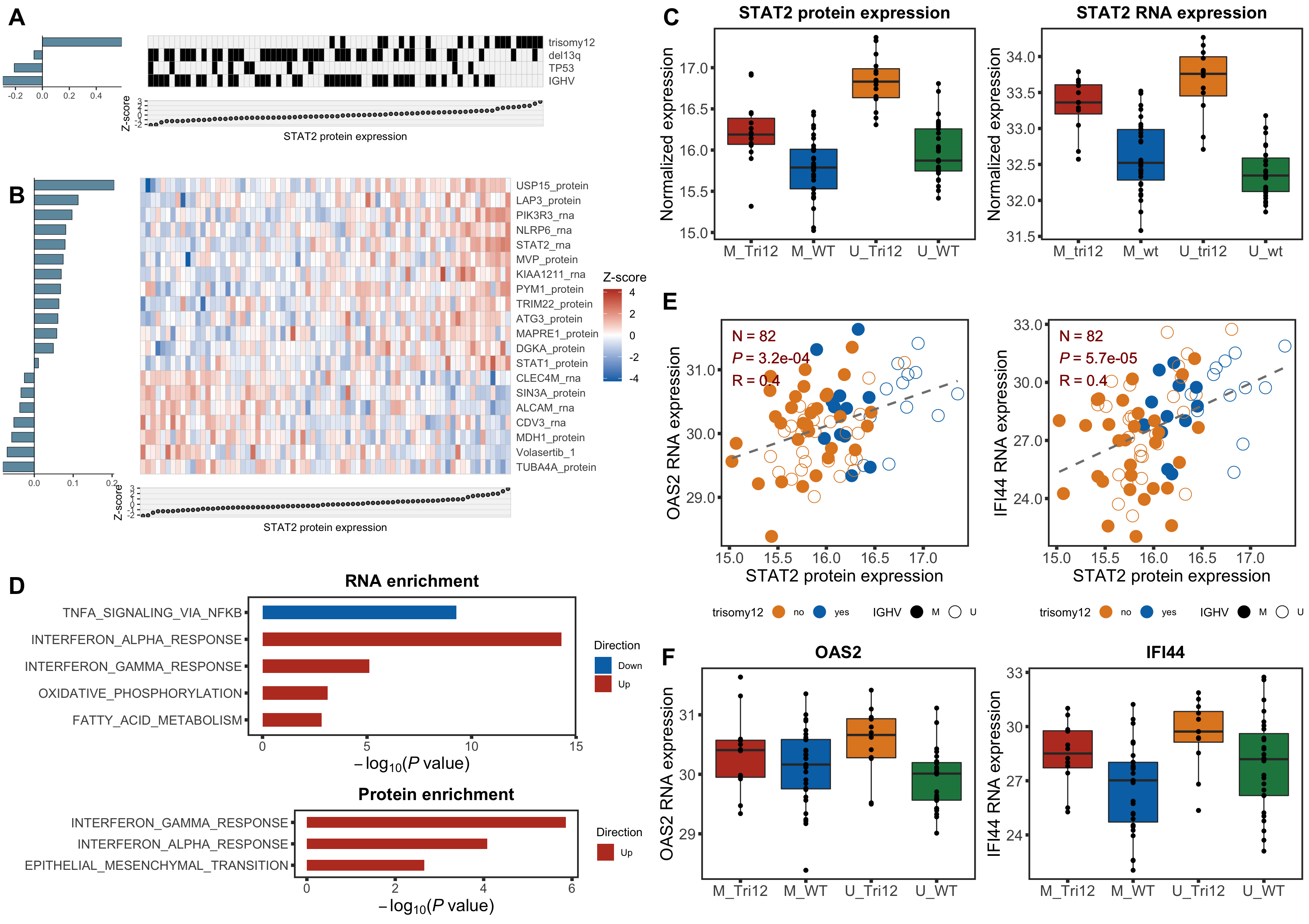
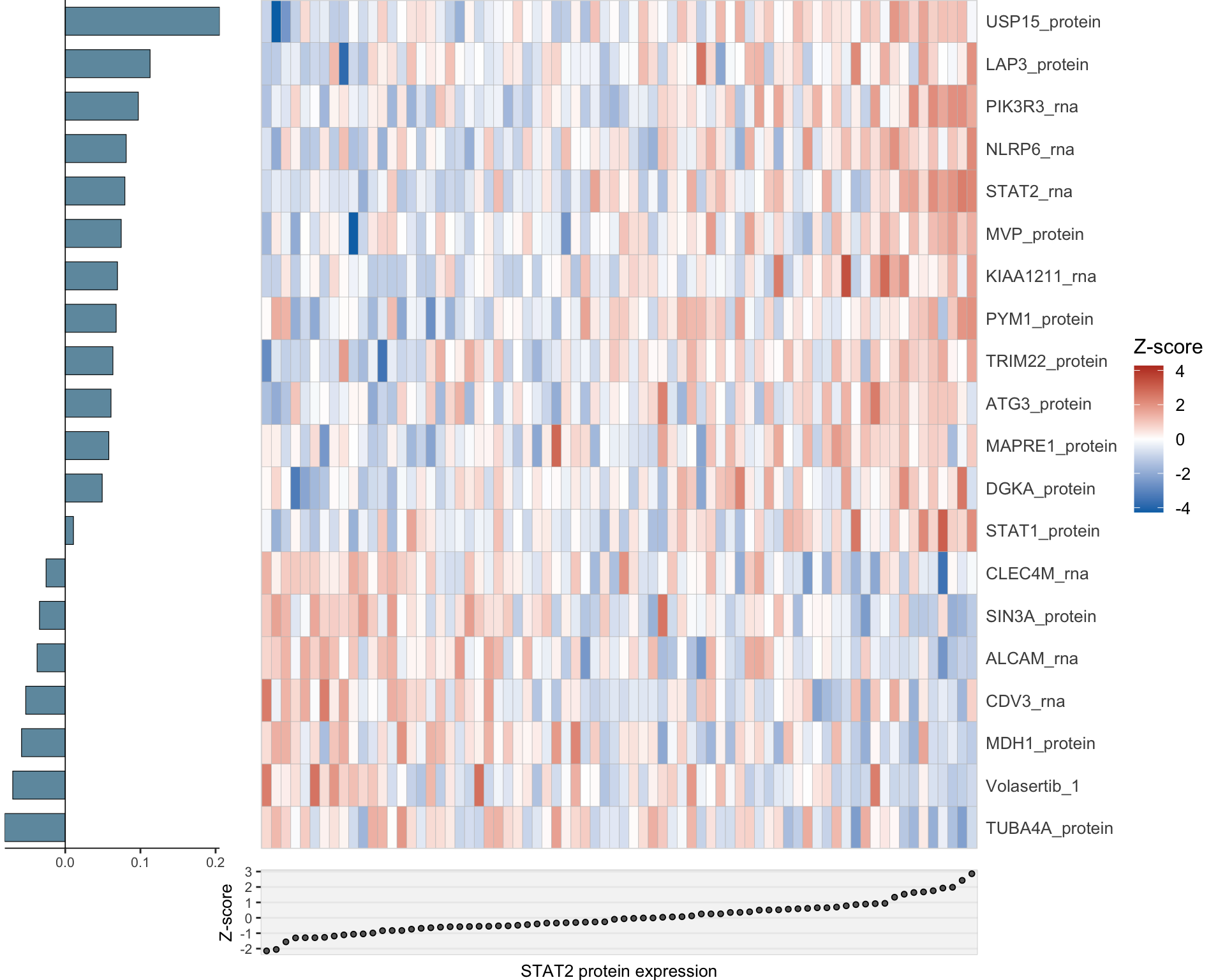
Variance explained for STAT2 expression by multi-omics datasets
Heatmap of selected features
library(gtable)
lassoPlot <- function(lassoOut, cleanData, freqCut = 1, coefCut = 0.01, setNumber = "last", legend = TRUE, labSuffix = " protein expression", scaleFac =1) {
plotList <- list()
if (setNumber == "last") {
setNumber <- length(lassoOut[[1]])
} else {
setNumber <- setNumber
}
for (seaName in names(lassoOut)) {
#for the barplot on the left of the heatmap
barValue <- rowMeans(lassoOut[[seaName]][[setNumber]]$coefMat)
freqValue <- rowMeans(abs(sign(lassoOut[[seaName]][[setNumber]]$coefMat)))
barValue <- barValue[abs(barValue) >= coefCut & freqValue >= freqCut] # a certain threshold
barValue <- barValue[order(barValue)]
if(length(barValue) == 0) {
plotList[[seaName]] <- NA
next
}
#for the heatmap and scatter plot below the heatmap
allData <- cleanData$allExplain[[seaName]][[setNumber]]
seaValue <- cleanData$allResponse[[seaName]]*2 #back to Z-score
tabValue <- allData[, names(barValue),drop=FALSE]
ord <- order(seaValue)
seaValue <- seaValue[ord]
tabValue <- tabValue[ord, ,drop=FALSE]
sampleIDs <- rownames(tabValue)
tabValue <- as.tibble(tabValue)
#change scaled binary back to catagorical
for (eachCol in colnames(tabValue)) {
if (strsplit(eachCol, split = "[.]")[[1]][1] != "con") {
tabValue[[eachCol]] <- as.integer(as.factor(tabValue[[eachCol]]))
}
else {
tabValue[[eachCol]] <- tabValue[[eachCol]]*2 #back to Z-score
}
}
tabValue$Sample <- sampleIDs
#Mark different rows for different scaling in heatmap
matValue <- gather(tabValue, key = "Var",value = "Value", -Sample)
matValue$Type <- "mut"
#For continuious value
matValue$Type[grep("con.",matValue$Var)] <- "con"
#for methylation_cluster
matValue$Type[grep("ConsCluster",matValue$Var)] <- "meth"
#change the scale of the value, let them do not overlap with each other
matValue[matValue$Type == "mut",]$Value = matValue[matValue$Type == "mut",]$Value + 10
matValue[matValue$Type == "meth",]$Value = matValue[matValue$Type == "meth",]$Value + 20
#color scale for viability
idx <- matValue$Type == "con"
myCol <- colorRampPalette(c(colList[2],'white',colList[1]),
space = "Lab")
if (sum(idx) != 0) {
matValue[idx,]$Value = round(matValue[idx,]$Value,digits = 2)
minViab <- min(matValue[idx,]$Value)
maxViab <- max(matValue[idx,]$Value)
limViab <- max(c(abs(minViab), abs(maxViab)))
scaleSeq1 <- round(seq(-limViab, limViab,0.01), digits=2)
color4viab <- setNames(myCol(length(scaleSeq1+1)), nm=scaleSeq1)
} else {
scaleSeq1 <- round(seq(0,1,0.01), digits=2)
color4viab <- setNames(myCol(length(scaleSeq1+1)), nm=scaleSeq1)
}
#change continues measurement to discrete measurement
matValue$Value <- factor(matValue$Value,levels = sort(unique(matValue$Value)))
#change order of heatmap
names(barValue) <- gsub("con.", "", names(barValue))
matValue$Var <- gsub("con.","",matValue$Var)
matValue$Var <- factor(matValue$Var, levels = names(barValue))
matValue$Sample <- factor(matValue$Sample, levels = names(seaValue))
#plot the heatmap
p1 <- ggplot(matValue, aes(x=Sample, y=Var)) + geom_tile(aes(fill=Value), color = "gray") +
theme_bw() + scale_y_discrete(expand=c(0,0),position = "right") +
theme(axis.text.y=element_text(hjust=0, size=10*scaleFac), axis.text.x=element_blank(),
axis.title = element_blank(),
axis.ticks=element_blank(), panel.border=element_rect(colour="gainsboro"),
plot.title=element_blank(), panel.background=element_blank(),
panel.grid.major=element_blank(), panel.grid.minor=element_blank(),
plot.margin = margin(0,0,0,0)) +
scale_fill_manual(name="Mutated", values=c(color4viab, `11`="gray96", `12`='black', `21`='lightgreen',
`22`='green',`23` = 'green4'),guide=FALSE) #+ ggtitle(seaName)
#Plot the bar plot on the left of the heatmap
barDF = data.frame(barValue, nm=factor(names(barValue),levels=names(barValue)))
p2 <- ggplot(data=barDF, aes(x=nm, y=barValue)) +
geom_bar(stat="identity", fill=colList[6], colour="black", position = "identity", width=.66, size=0.2) +
theme_bw() + geom_hline(yintercept=0, size=0.3) + scale_x_discrete(expand=c(0,0.5)) +
scale_y_continuous(expand=c(0,0)) + coord_flip() +
theme(panel.grid.major=element_blank(), panel.background=element_blank(), axis.ticks.y = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x =element_text(size=8*scaleFac),
axis.text.y = element_blank(),
axis.title = element_blank(),
panel.border=element_blank(),plot.margin = margin(0,0,0,0)) + geom_vline(xintercept=c(0.5), color="black", size=0.6)
#Plot the scatter plot under the heatmap
# scatterplot below
scatterDF = data.frame(X=factor(names(seaValue), levels=names(seaValue)), Y=seaValue)
p3 <- ggplot(scatterDF, aes(x=X, y=Y)) + geom_point(shape=21, fill="dimgrey", colour="black", size=1.2) +
xlab(paste0(seaName, labSuffix)) + ylab("Z-score") +
theme_bw() +
theme(panel.grid.minor=element_blank(), panel.grid.major.x=element_blank(),
axis.title=element_text(size=10*scaleFac),
axis.text.x=element_blank(), axis.ticks.x=element_blank(),
axis.text.y=element_text(size=8*scaleFac),
panel.border=element_rect(colour="dimgrey", size=0.1),
panel.background=element_rect(fill="gray96"),plot.margin = margin(0,0,0,0))
dummyGrob <- ggplot() + theme_void()
#Scale bar for continuous variable
if (legend) {
Vgg = ggplot(data=data.frame(x=1, y=as.numeric(names(color4viab))), aes(x=x, y=y, color=y)) + geom_point() +
scale_color_gradientn(name="Z-score", colours =color4viab) +
theme(legend.title=element_text(size=12*scaleFac), legend.text=element_text(size=10*scaleFac))
barLegend <- plot_grid(gtable_filter(ggplotGrob(Vgg), "guide-box"))
#Assemble all the plots togehter
} else {
barLegend <- dummyGrob
}
gt <- egg::ggarrange(p2,p1,barLegend,dummyGrob, p3, dummyGrob, ncol=3, nrow=2,
widths = c(0.6,2,0.3), padding = unit(0,"line"), clip = "off",
heights = c(length(unique(matValue$Var))/2,1),draw = FALSE)
plotList[[seaName]] <- gt
}
return(plotList)
}
Genetics only
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 1,setNumber = 1, legend = FALSE, scaleFac = 1)
heatMaps <- heatMaps[!is.na(heatMaps)]
geneLasso <- plot_grid(plotlist=heatMaps, ncol=1)
geneLasso
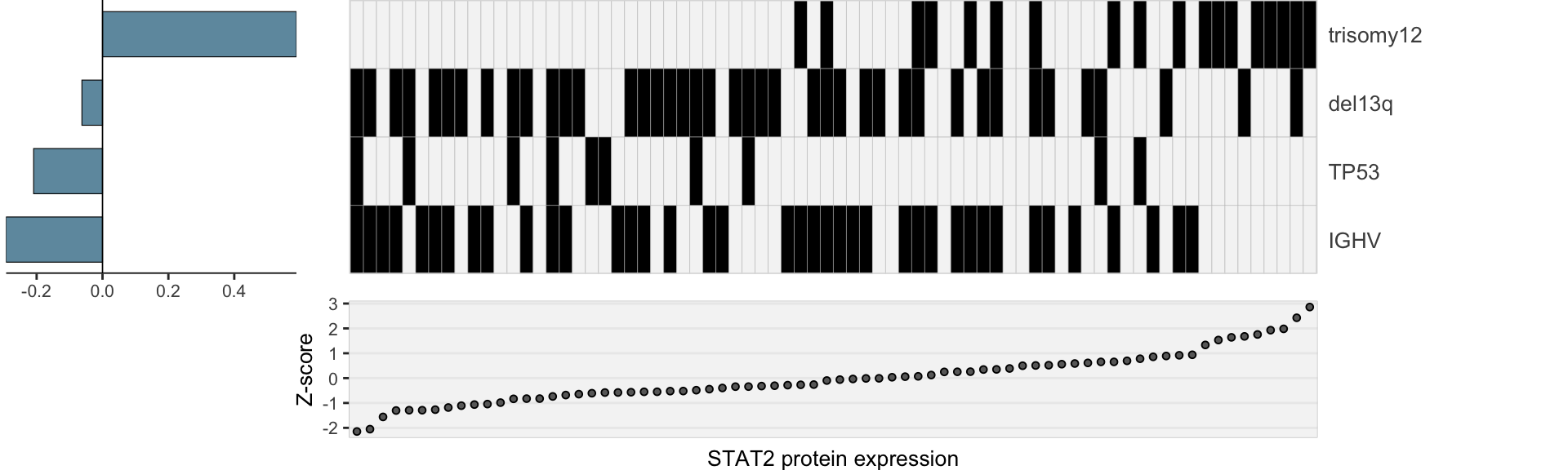
Combined
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 1,setNumber = 5, legend = TRUE, scaleFac = 1)
heatMaps <- heatMaps[!is.na(heatMaps)]
comLasso <- plot_grid(plotlist=heatMaps, ncol=1)
comLasso
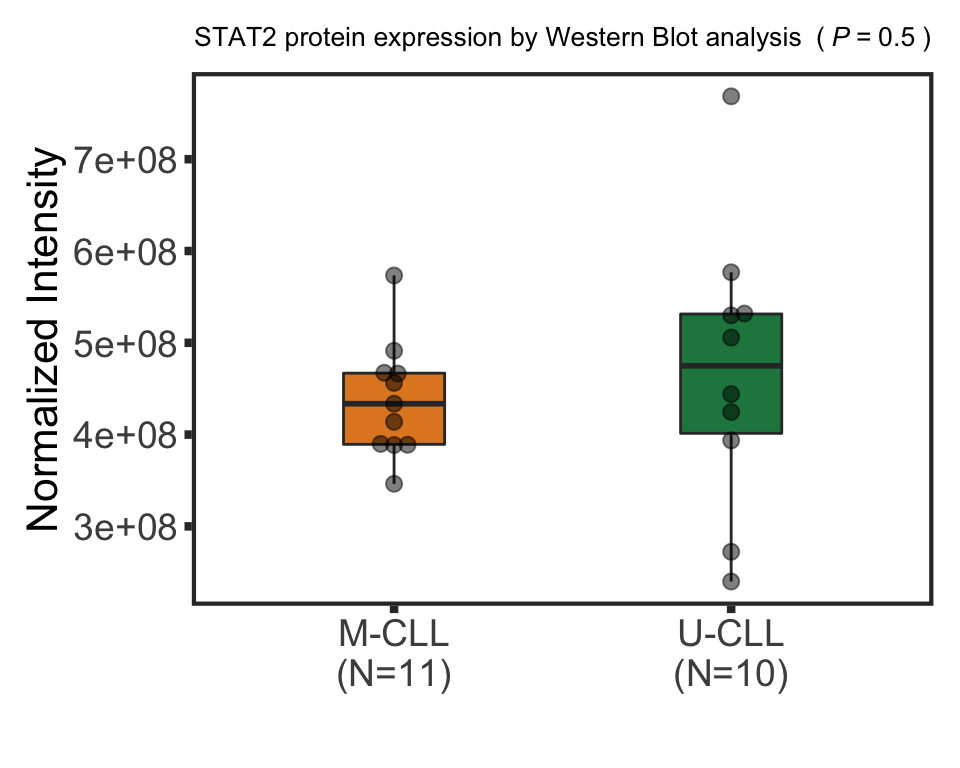
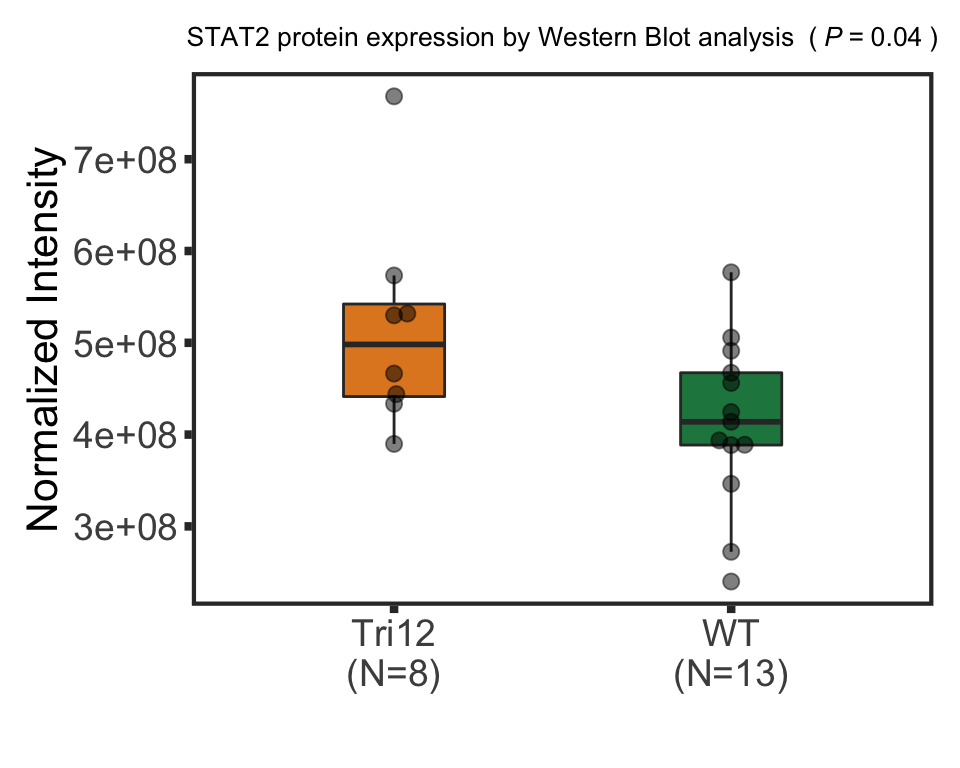
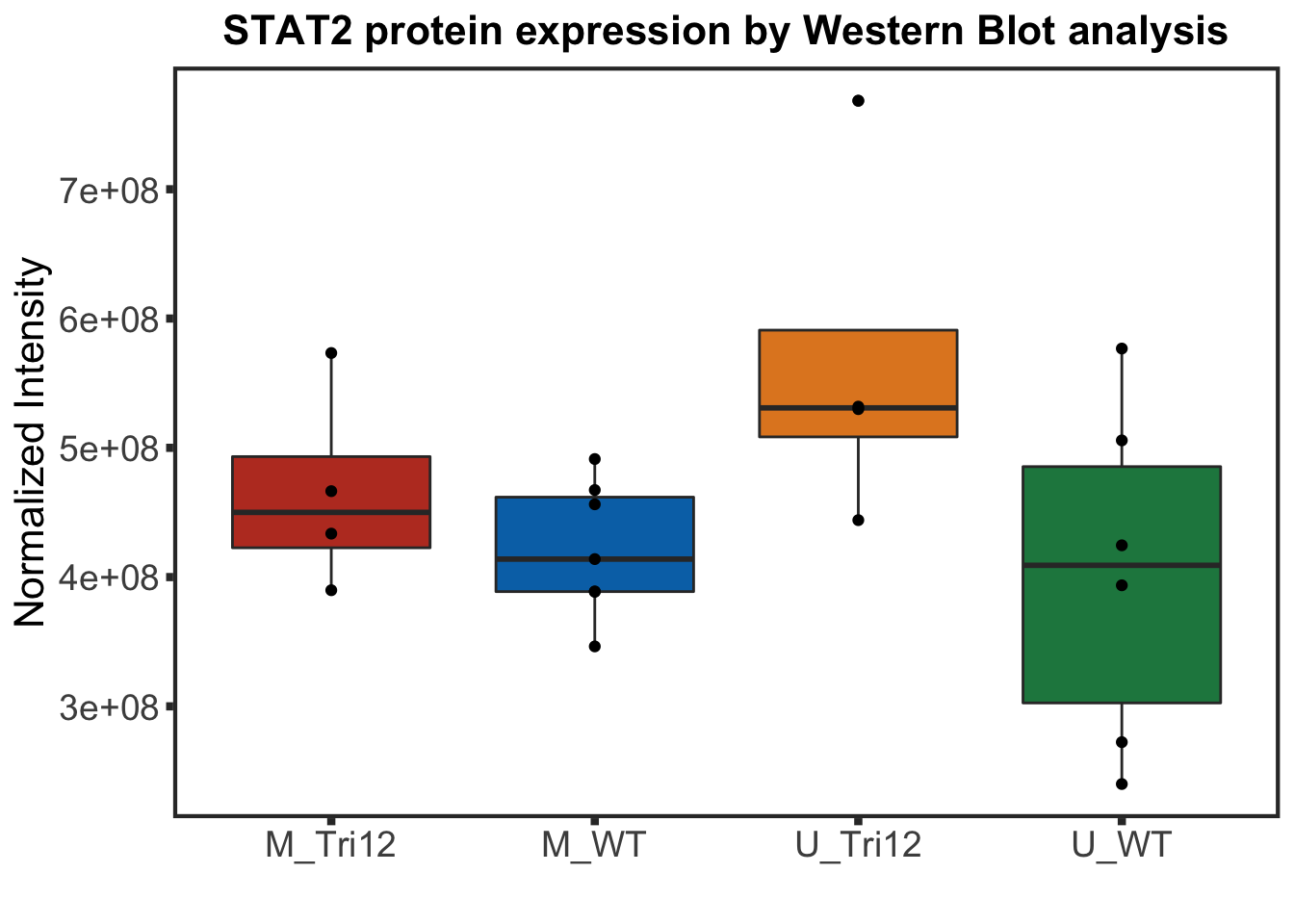
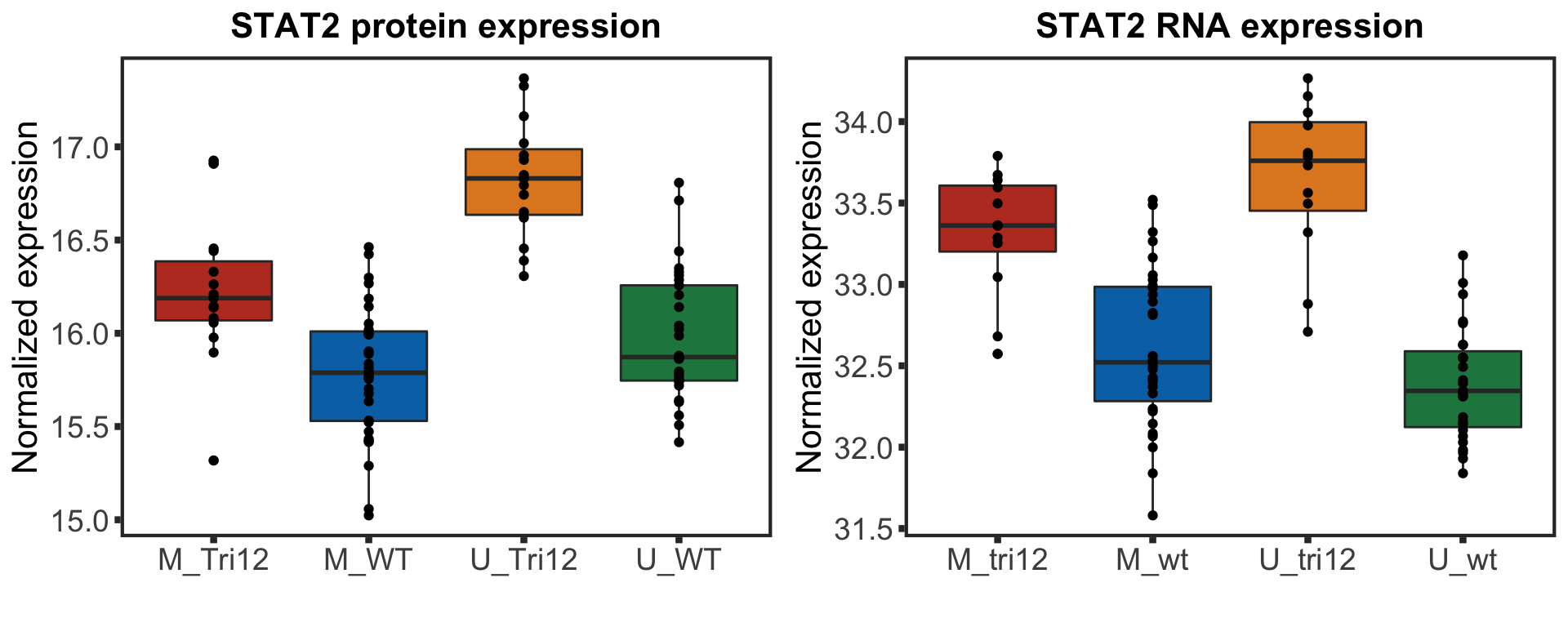
STAT2 protein expression stratified by IGHV and trisomy12
plotTab <- tibble(patID = colnames(protCLL),
STAT2 = assays(protCLL)[["count_combat"]][rowData(protCLL)$hgnc_symbol == "STAT2",],
trisomy12 = protCLL$trisomy12,
IGHV=protCLL$IGHV.status) %>%
filter(!is.na(IGHV), !is.na(trisomy12)) %>%
mutate(trisomy12 = ifelse(trisomy12 == 0, "WT","Tri12")) %>%
mutate(group = paste0(IGHV, "_", trisomy12))
stat2BoxProt <- ggplot(plotTab, aes(group, y=STAT2, fill = group)) + geom_boxplot() + geom_point() + theme_full +
scale_fill_manual(values = colList) + theme(legend.position = "none") +
xlab("") + ggtitle("STAT2 protein expression") +ylab("Normalized expression")
summary(lm(STAT2 ~ IGHV * trisomy12, plotTab))
Call:
lm(formula = STAT2 ~ IGHV * trisomy12, data = plotTab)
Residuals:
Min 1Q Median 3Q Max
-0.9044 -0.2317 -0.0275 0.2222 0.8272
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 16.22239 0.09137 177.538 < 2e-16 ***
IGHVU 0.60433 0.12922 4.677 1.06e-05 ***
trisomy12WT -0.43065 0.11074 -3.889 0.000196 ***
IGHVU:trisomy12WT -0.41597 0.15789 -2.634 0.009974 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.3539 on 87 degrees of freedom
Multiple R-squared: 0.5157, Adjusted R-squared: 0.499
F-statistic: 30.87 on 3 and 87 DF, p-value: 1.097e-13
STAT2 protein expression is strongly affected by IGHV and trisomy12 status, U-CLLs with trisomy12 shows significant up-regulation of STAT2
STAT2 RNA expression stratified by IGHV and trisomy12
Samples in the proteomic cohort
plotTab <- tibble(patID = colnames(ddsSub.voom),
STAT2 = assay(ddsSub.voom)[rowData(ddsSub.voom)$symbol == "STAT2",],
trisomy12 = patMeta[match(patID, patMeta$Patient.ID),]$trisomy12,
IGHV=patMeta[match(patID, patMeta$Patient.ID),]$IGHV.status) %>%
mutate(trisomy12 = ifelse(trisomy12 == 0, "wt","tri12")) %>%
mutate(group = paste0(IGHV, "_", trisomy12))
stat2BoxRNA <- ggplot(plotTab, aes(group, y=STAT2, fill = group)) + geom_boxplot() + geom_point() + theme_full +
scale_fill_manual(values = colList) + theme(legend.position = "none") +
xlab("") + ggtitle("STAT2 RNA expression") +ylab("Normalized expression")
summary(lm(STAT2 ~ IGHV * trisomy12, plotTab))
Call:
lm(formula = STAT2 ~ IGHV * trisomy12, data = plotTab)
Residuals:
Min 1Q Median 3Q Max
-1.03193 -0.27905 -0.03574 0.32709 0.90706
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 33.3132 0.1245 267.644 < 2e-16 ***
IGHVU 0.3326 0.1760 1.889 0.06257 .
trisomy12wt -0.7005 0.1466 -4.778 8.16e-06 ***
IGHVU:trisomy12wt -0.5541 0.2094 -2.646 0.00986 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.4312 on 78 degrees of freedom
Multiple R-squared: 0.5446, Adjusted R-squared: 0.5271
F-statistic: 31.09 on 3 and 78 DF, p-value: 2.525e-13
stat2Box <- plot_grid(stat2BoxProt, stat2BoxRNA)
stat2Box
Pathway enrichment for RNA expressions correlated with STAT2 protein expression
expVar <- "STAT2"
protMat <- assays(protCLL)[["QRILC_combat"]]
rownames(protMat) <- rowData(protCLL)$hgnc_symbol
yVec <- protMat[expVar,]
Prepare data
#subset
ddsSub <- dds[,dds$PatID %in% names(yVec)]
#only keep protein coding genes with symbol
ddsSub <- ddsSub[rowData(ddsSub)$biotype %in% "protein_coding" & !rowData(ddsSub)$symbol %in% c("",NA),]
#remove lowly expressed genes
ddsSub <- ddsSub[rowSums(counts(ddsSub, normalized = TRUE)) > 100,]
#voom transformation
exprMat <- limma::voom(counts(ddsSub), lib.size = ddsSub$sizeFactor)$E
ddsSub.voom <- ddsSub
assay(ddsSub.voom) <- exprMat
rnaMat <- exprMat
rownames(rnaMat) <- rowData(ddsSub.voom)$symbol
overSampe <- intersect(names(yVec), colnames(rnaMat))
rnaMat <- rnaMat[,overSampe]
yVec <- yVec[overSampe]
Test
#buid design matirx
ighv <- patMeta[match(names(yVec),patMeta$Patient.ID),]$IGHV.status
tri12 <- patMeta[match(names(yVec),patMeta$Patient.ID),]$trisomy12
d0 <- model.matrix(~yVec)
d1 <- model.matrix(~ighv+tri12+yVec)
no blocking for IGHV or trisomy12
fit <- lmFit(rnaMat, design = d0)
fit2 <- eBayes(fit)
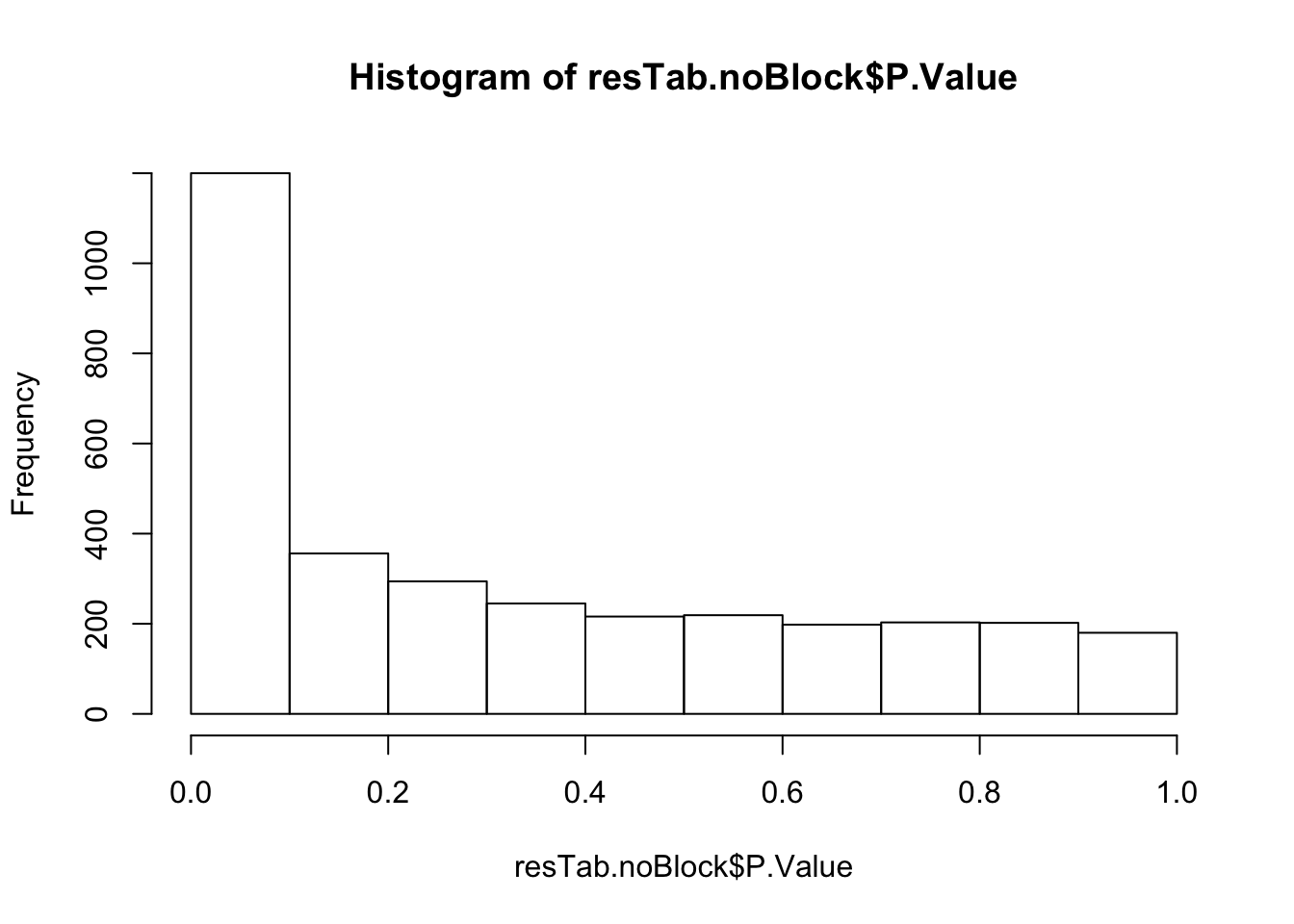
resTab.noBlock <- topTable(fit2, number = Inf, coef = "yVec") %>% data.frame() %>% rownames_to_column("name")
hist(resTab.noBlock$P.Value)
resTab.noBlock.sig <- filter(resTab.noBlock, adj.P.Val < 0.1)
resTab.noBlock %>% mutate_if(is.numeric, formatC, digits=2, format="e") %>% DT::datatable()
plotCorScatter <- function(plotTab, x_lab = "X", y_lab = "Y", title = "",
showR2 = FALSE, annoPos = "right",
dotCol = colList, textCol="darkred") {
#prepare annotation values
corRes <- cor.test(plotTab$x, plotTab$y)
pval <- formatNum(corRes$p.value, digits = 1, format = "e")
Rval <- formatNum(corRes$estimate, digits = 1, format = "e")
R2val <- formatNum(corRes$estimate^2, digits = 1, format = "e")
Nval <- nrow(plotTab)
annoP <- bquote(italic("P")~"="~.(pval))
if (showR2) {
annoCoef <- bquote(R^2~"="~.(R2val))
} else {
annoCoef <- bquote(R~"="~.(Rval))
}
annoN <- bquote(N~"="~.(Nval))
corPlot <- ggplot(plotTab, aes(x = x, y = y)) + geom_point(aes(col = trisomy12, shape = IGHV), size=5) +
scale_shape_manual(values = c(M = 19, U = 1)) +
scale_color_manual(values = c(yes = colList[2], no = colList[3])) +
geom_smooth(formula = y~x,method = "lm", se=FALSE, color = "grey50", linetype ="dashed" )
if (annoPos == "right") {
corPlot <- corPlot + annotate("text", x = max(plotTab$x), y = Inf, label = annoN,
hjust=1, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$x), y = Inf, label = annoP,
hjust=1, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$x), y = Inf, label = annoCoef,
hjust=1, vjust =6, size = 5, parse = FALSE, col= textCol)
} else if (annoPos== "left") {
corPlot <- corPlot + annotate("text", x = min(plotTab$x), y = Inf, label = annoN,
hjust=0, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$x), y = Inf, label = annoP,
hjust=0, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$x), y = Inf, label = annoCoef,
hjust=0, vjust =6, size = 5, parse = FALSE, col= textCol)
}
corPlot <- corPlot + ylab(y_lab) + xlab(x_lab) + ggtitle(title) +
scale_y_continuous(labels = scales::number_format(accuracy = 0.1)) +
scale_x_continuous(labels = scales::number_format(accuracy = 0.1)) +
theme_full + theme(legend.position = "bottom", plot.margin = margin(12,12,12,12))
corPlot
}
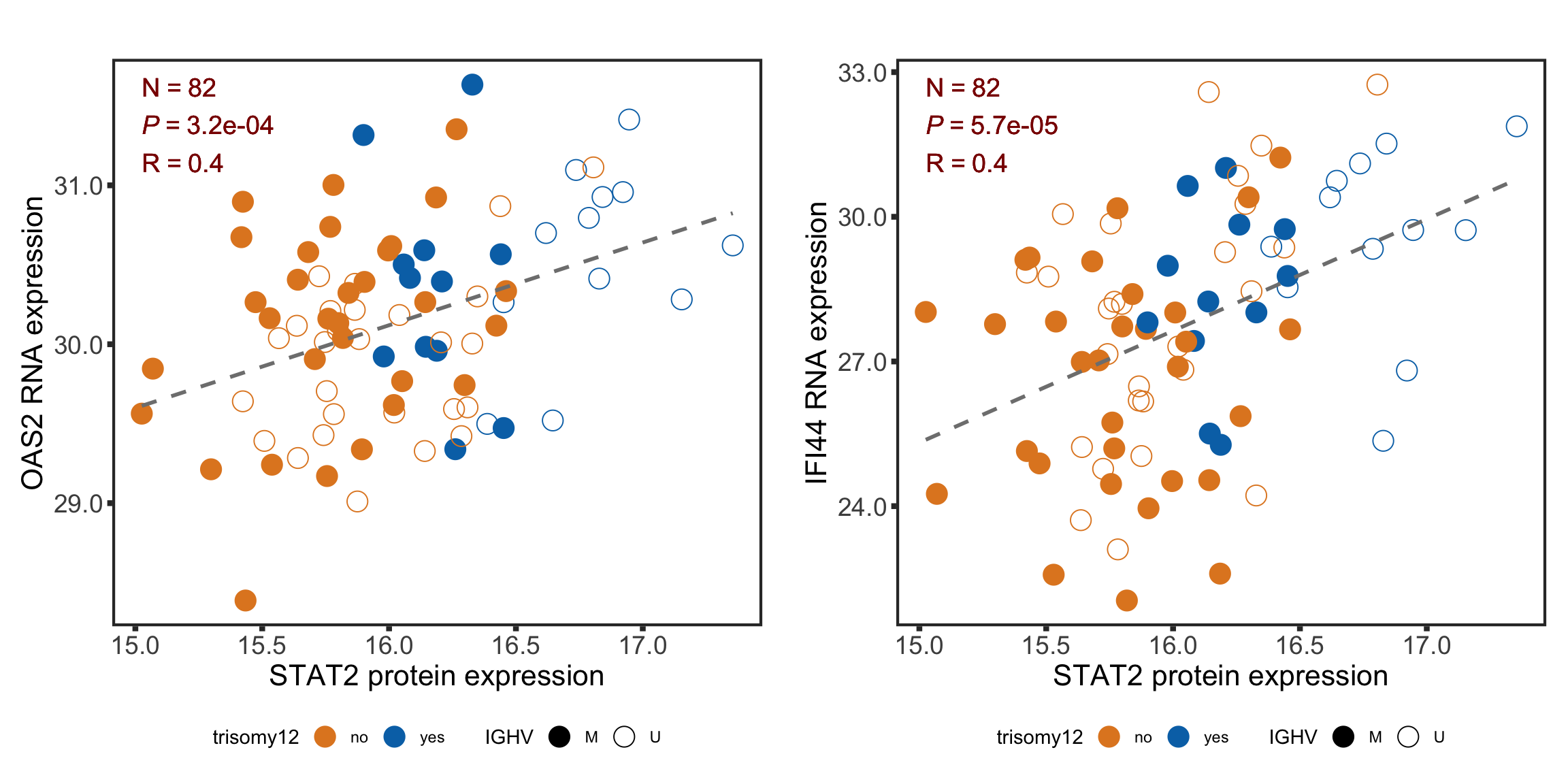
Correlation between selected genes and STAT2 protein expression
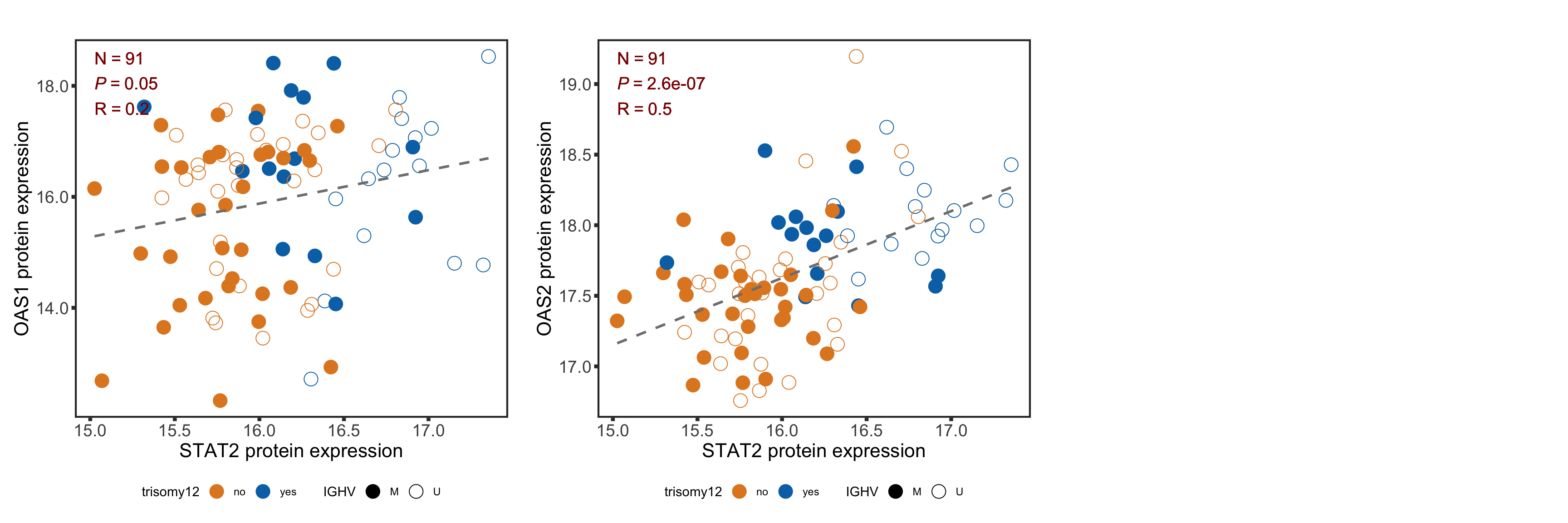
geneList <- c("OAS2", "IFI44")
rnaSTAT2cor <- lapply(geneList, function(n) {
plotTab <- tibble(x = yVec, y = rnaMat[n,], IGHV = ighv, tri12 = tri12) %>%
mutate(trisomy12 = ifelse(tri12==1,"yes","no"))
plotCorScatter(plotTab, annoPos = "left", x_lab = "STAT2 protein expression", y_lab = sprintf("%s RNA expression", n))
})
names(rnaSTAT2cor) <- geneList
plotRNAcor <- plot_grid(plotlist = rnaSTAT2cor)
plotRNAcor
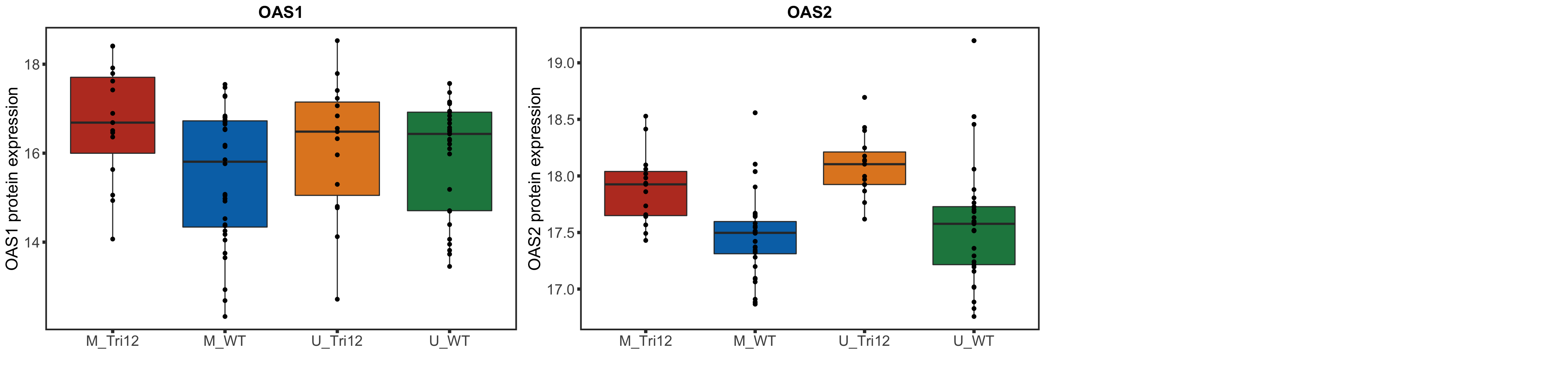
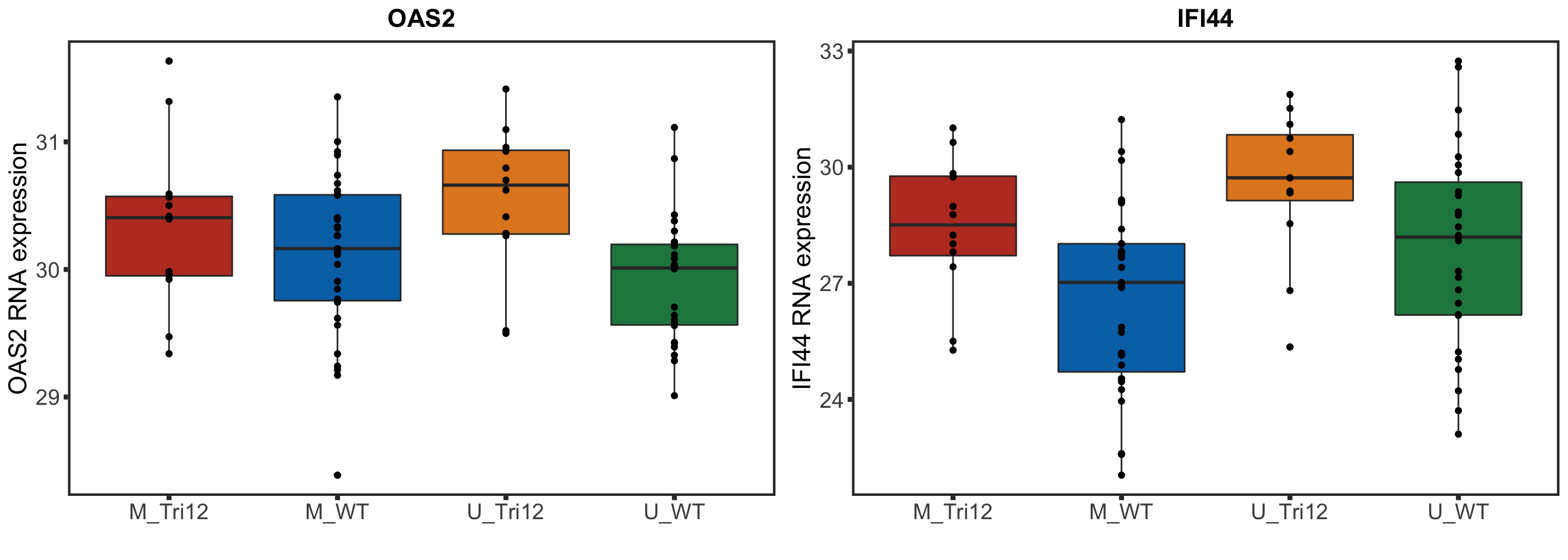
geneList <- c("OAS2", "IFI44")
rnaSTAT2corBox <- lapply(geneList, function(n) {
plotTab <- tibble(expr = rnaMat[n,], IGHV = ighv, tri12 = tri12) %>%
mutate(trisomy12 = ifelse(tri12==1,"Tri12","WT")) %>%
mutate(group = paste0(IGHV, "_", trisomy12))
ggplot(plotTab, aes(group, y=expr, fill = group)) + geom_boxplot() + geom_point() + theme_full +
scale_fill_manual(values = colList) + theme(legend.position = "none") +
xlab("") + ylab(sprintf("%s RNA expression",n)) + ggtitle(n)
})
names(rnaSTAT2corBox) <- geneList
plotRNAbox <- plot_grid(plotlist = rnaSTAT2corBox)
plotRNAbox
gmts <- list(H = "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
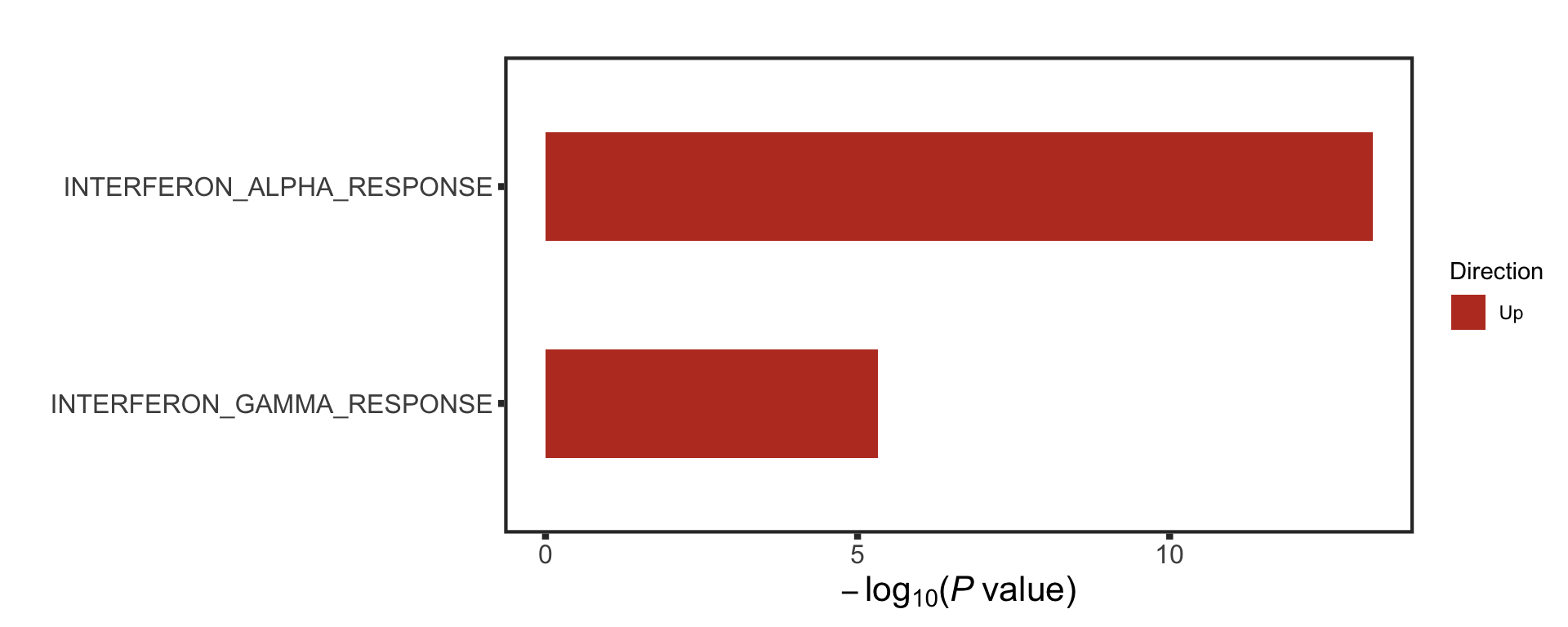
enRes <- runCamera(rnaMat, d0, gmts$H,
removePrefix = "HALLMARK_", pCut = 0.1, ifFDR = TRUE)
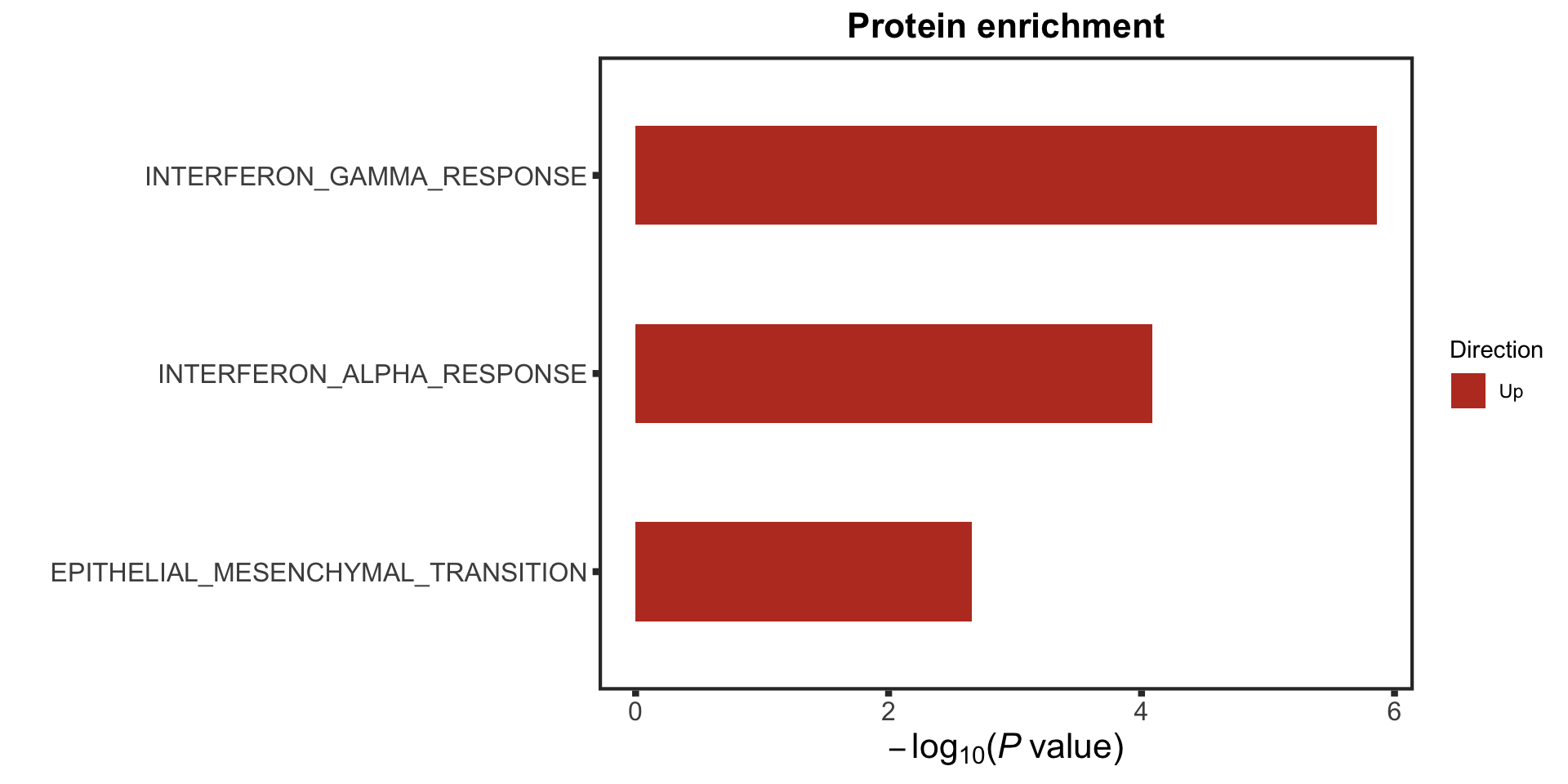
enRes$enrichPlot
colAnno <- tibble(id = names(yVec), STAT2= yVec, IGHV = ighv, trisomy12 = tri12) %>%
mutate(trisomy12 = ifelse(trisomy12 ==1,"yes","no")) %>%
data.frame() %>% column_to_rownames("id")
annoCol <- list(trisomy12 = c(yes = "black",no = "grey80"),
IGHV = c(M = colList[4], U = colList[3]),
STAT2 = circlize::colorRamp2(c(min(colAnno$STAT2),max(colAnno$STAT2)),
c("white", "green")))
nameList <- c("STAT2","IFI44","OAS1","OAS2", "IFI30")
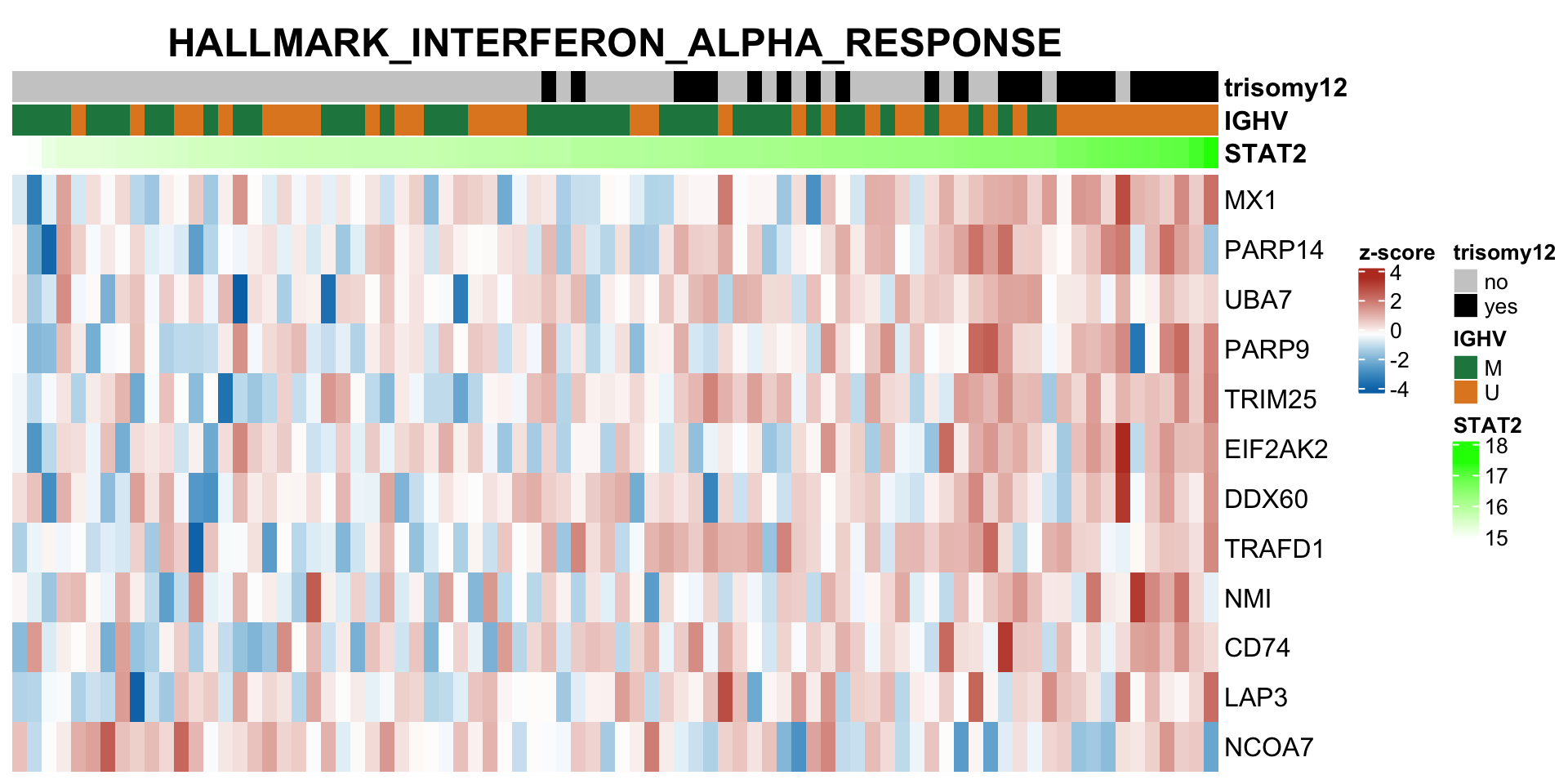
plotSetHeatmap(resTab.noBlock.sig, gmts$H, "HALLMARK_INTERFERON_ALPHA_RESPONSE", rnaMat, colAnno = colAnno, annoCol = annoCol, highLight = nameList)
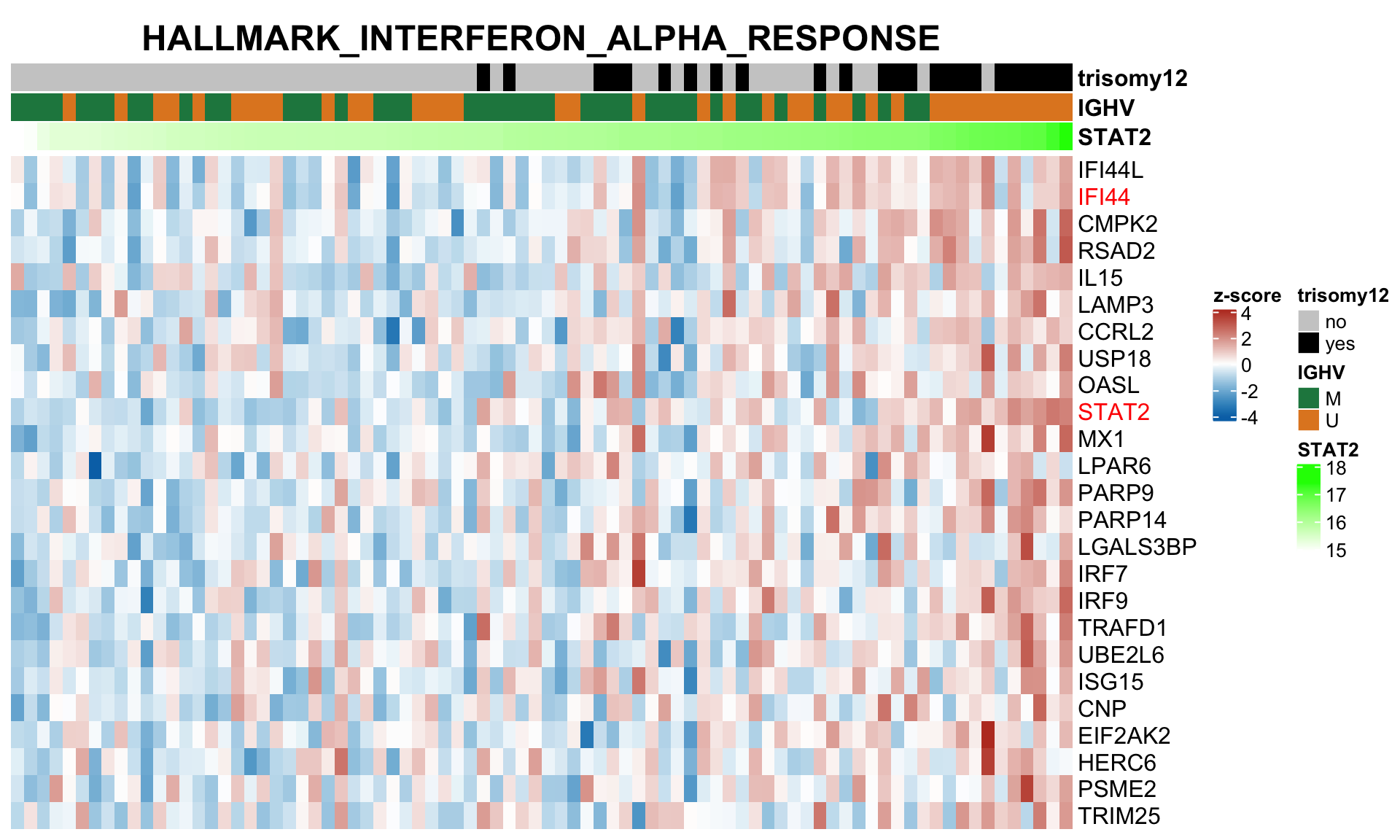
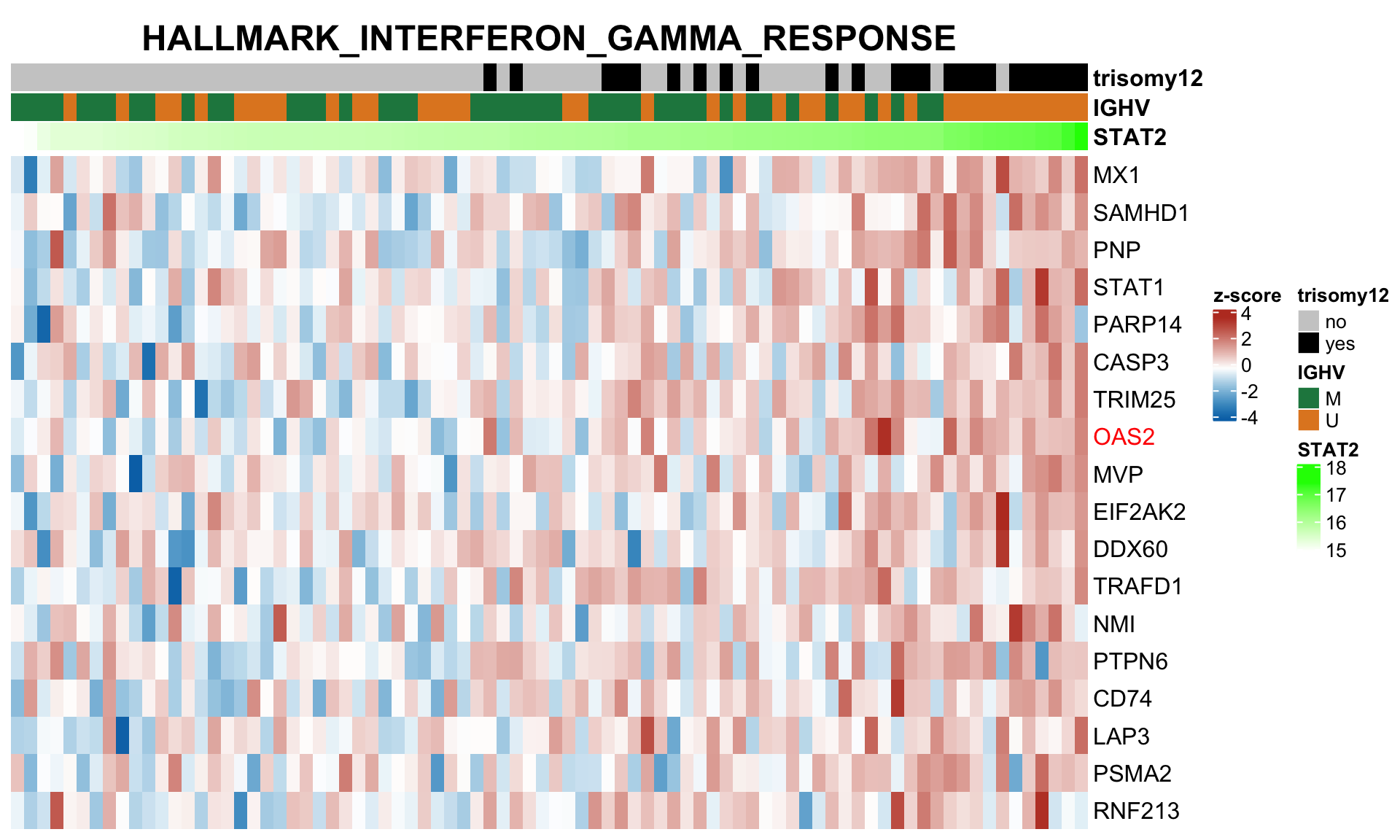
plotSetHeatmap(resTab.noBlock.sig, gmts$H, "HALLMARK_INTERFERON_GAMMA_RESPONSE", rnaMat, colAnno = colAnno, annoCol = annoCol,highLight = nameList)
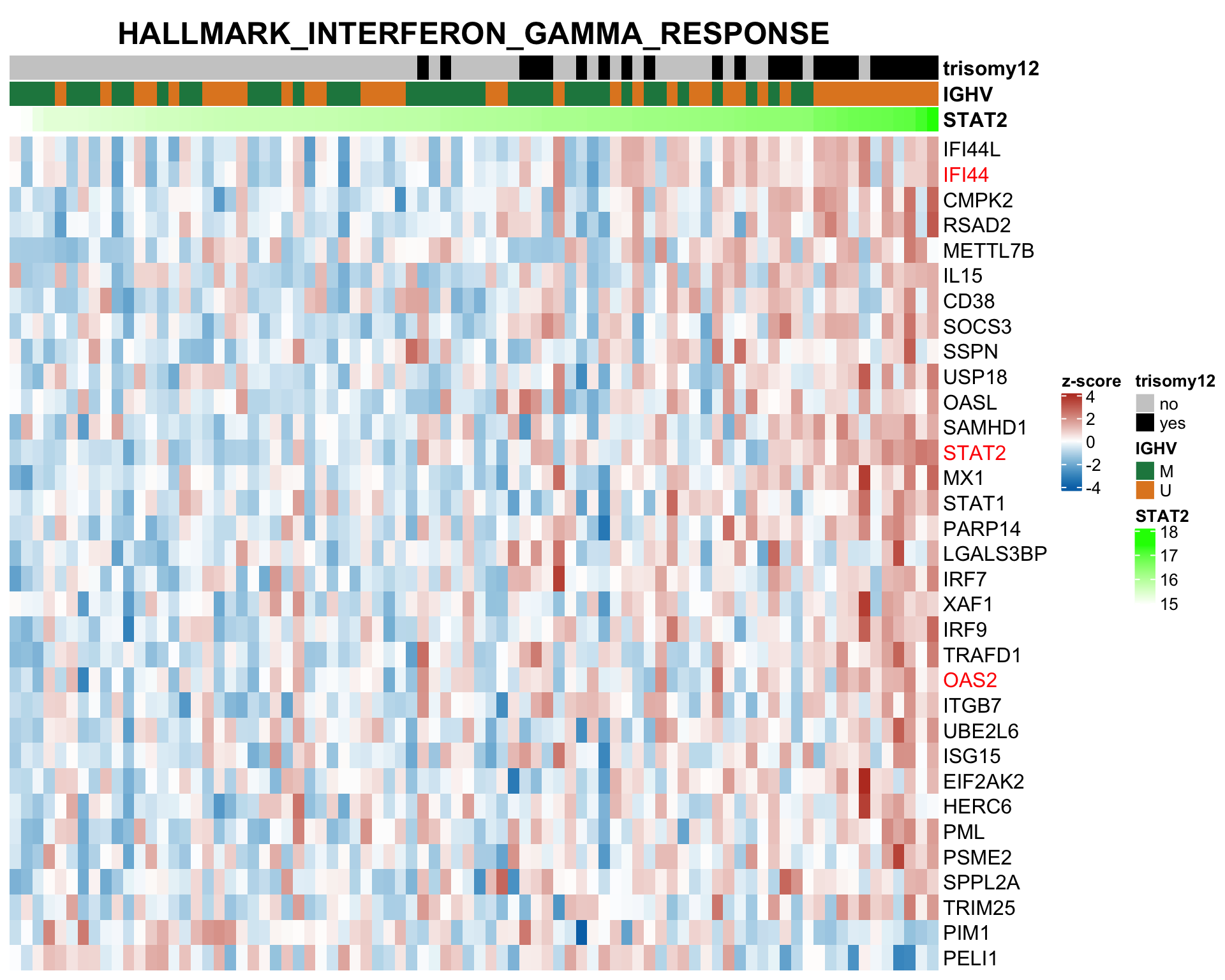
blocking for IGHV and trisomy12
fit <- lmFit(rnaMat, design = d1)
fit2 <- eBayes(fit)
resTab.block <- topTable(fit2, number = Inf, coef = "yVec") %>% data.frame() %>% rownames_to_column("name")
hist(resTab.block$P.Value)
resTab.block.sig <- filter(resTab.block, P.Value < 0.01)
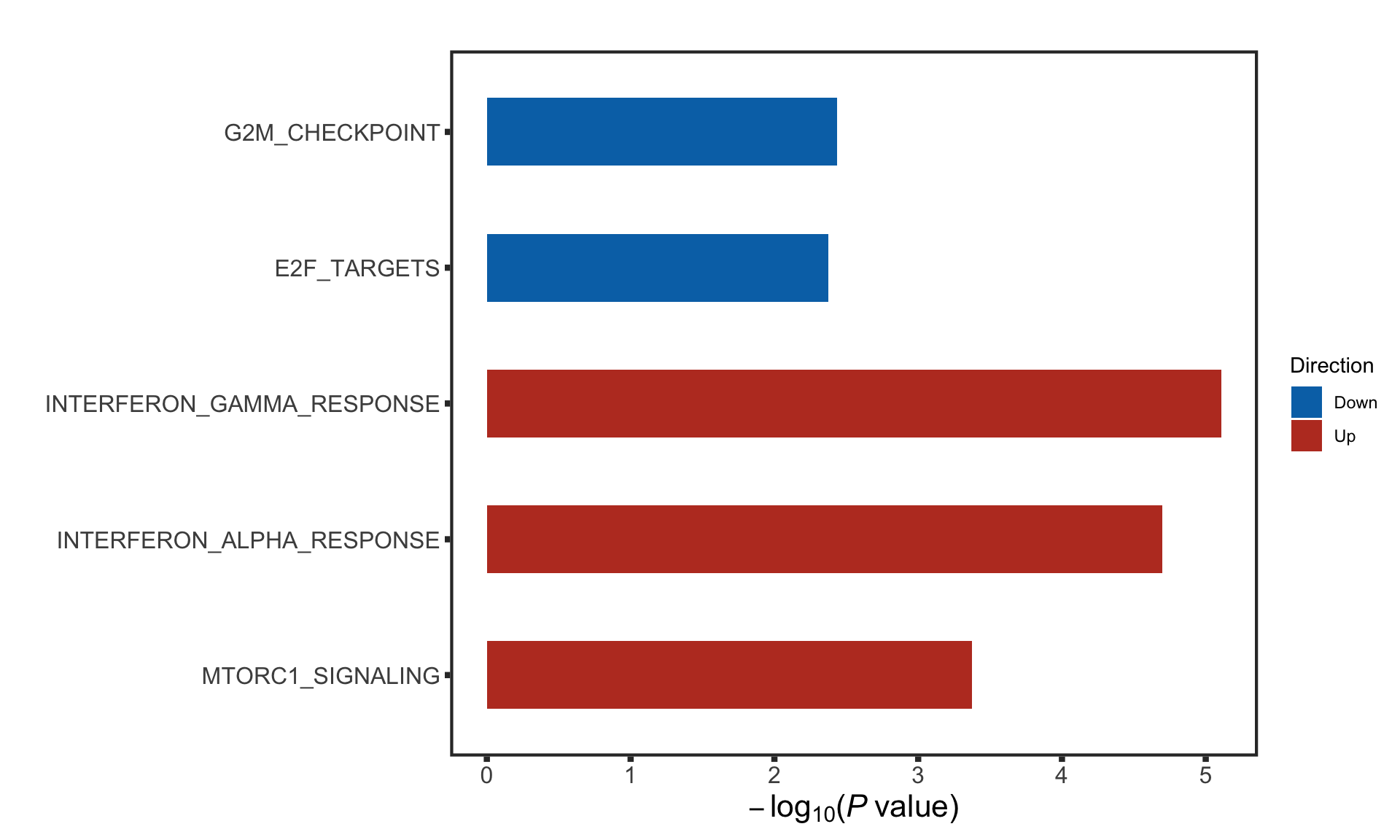
enRes <- runCamera(rnaMat, d1, gmts$H, contrast = "yVec",
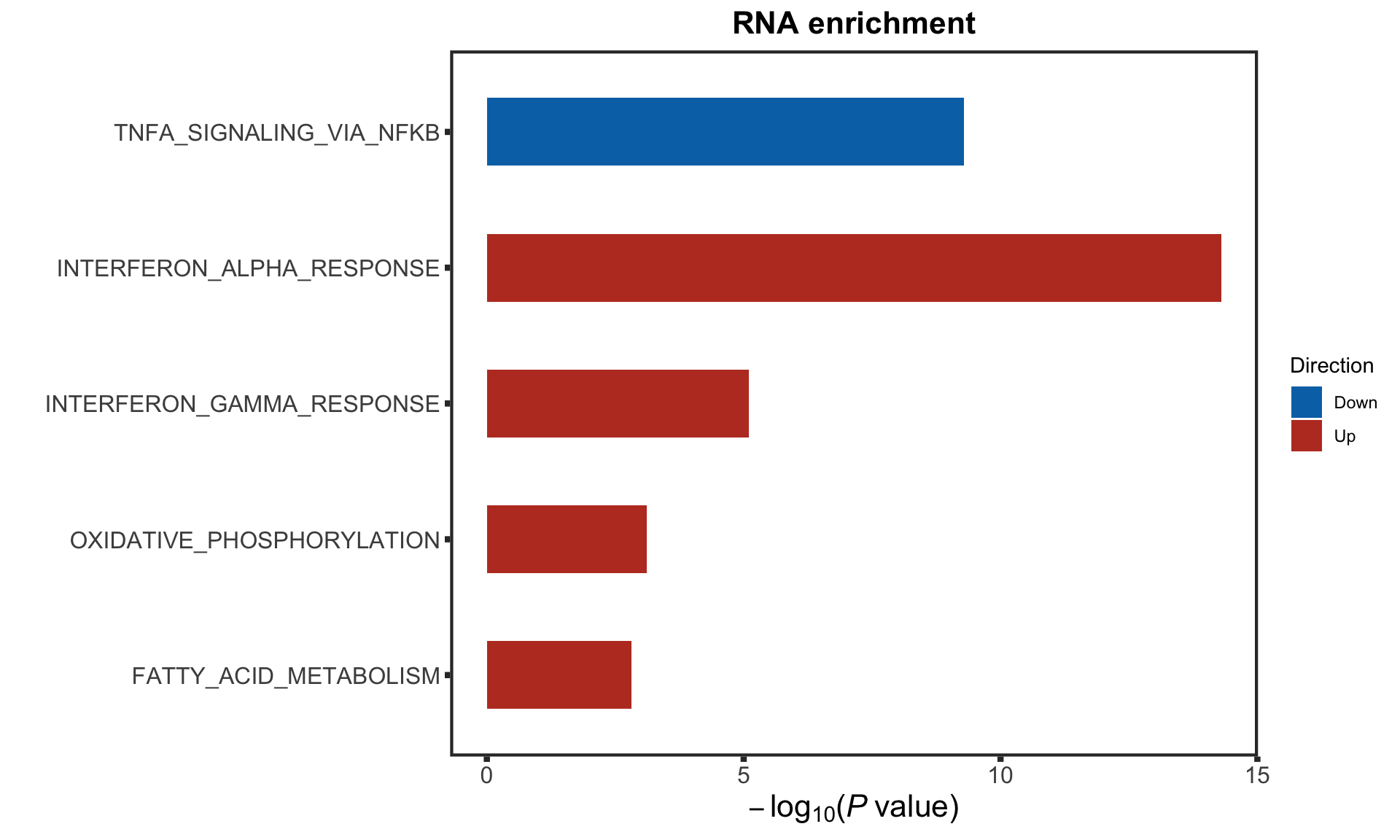
removePrefix = "HALLMARK_", pCut = 0.05, ifFDR = TRUE, plotTitle = "RNA enrichment")
rnaEnrich <- enRes$enrichPlot
rnaEnrich