Protein complex analysis of IGHV (an alternative approach)
Junyan Lu
2020-05-22
Last updated: 2020-06-16
Checks: 5 2
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- unnamed-chunk-5
To ensure reproducibility of the results, delete the cache directory complexAnalysis_IGHV_alternative_cache and re-run the analysis. To have workflowr automatically delete the cache directory prior to building the file, set delete_cache = TRUE when running wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/analysisDrugResponses_IC50_cache/
Ignored: analysis/analysisDrugResponses_cache/
Ignored: analysis/complexAnalysis_IGHV_alternative_cache/
Ignored: analysis/complexAnalysis_IGHV_cache/
Ignored: analysis/complexAnalysis_trisomy12_alteredPQR_cache/
Ignored: analysis/complexAnalysis_trisomy12_alternative_cache/
Ignored: analysis/complexAnalysis_trisomy12_cache/
Ignored: analysis/correlateCLLPD_cache/
Ignored: analysis/predictOutcome_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/CNVanalysis_11q.Rmd
Untracked: analysis/CNVanalysis_trisomy12.Rmd
Untracked: analysis/CNVanalysis_trisomy19.Rmd
Untracked: analysis/analysisDrugResponses.Rmd
Untracked: analysis/analysisDrugResponses_IC50.Rmd
Untracked: analysis/analysisPCA.Rmd
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/complexAnalysis_IGHV.Rmd
Untracked: analysis/complexAnalysis_IGHV_alternative.Rmd
Untracked: analysis/complexAnalysis_overall.Rmd
Untracked: analysis/complexAnalysis_trisomy12.Rmd
Untracked: analysis/complexAnalysis_trisomy12_alternative.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/correlateRNAexpression.Rmd
Untracked: analysis/default.css
Untracked: analysis/del11q.pdf
Untracked: analysis/del11q_norm.pdf
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/plotExpressionCNV.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/style.css
Untracked: analysis/trisomy12.pdf
Untracked: analysis/trisomy12_AFcor.Rmd
Untracked: analysis/trisomy12_norm.pdf
Untracked: code/AlteredPQR.R
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/example_proteomics_data
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/proteins_in_complexes
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/LUMOS_processed.RData
Untracked: output/cnv_plots.zip
Untracked: output/cnv_plots/
Untracked: output/cnv_plots_norm.zip
Untracked: output/dxdCLL.RData
Untracked: output/exprCNV.RData
Untracked: output/lassoResults_CPS.RData
Untracked: output/lassoResults_IC50.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/plotCNV_allChr11_diff.pdf
Untracked: output/plotCNV_del11q_sum.pdf
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_LUMOS_20200430.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/splicingResults.RData
Untracked: output/timsTOF_processed.RData
Untracked: plotCNV_del11q_diff.pdf
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateCLLPD.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Goal
In this analysis, I am trying to answer the question that how IGHV mutation status affect protein abundance through protein complexes. As the gene dosage effect is not involved, it’s hard to define a causal effect. I will define the proteins that show both RNA and protein levels changes related to IGHV as “cause”, which may be a more direct impact by IGHV (similar to effect of trisomy12 on the proteins from chr12). On the other hand, I will define the proteins that only show protein levels changes but not RNA level changes as “effect”, in which the protein abundance changes can be explained by complex formation but not RNA expression. Then the analysis steps are similar to the trisomy12 analysis
Analysis
Load libraries and dataset
library(SummarizedExperiment)
library(tidygraph)
library(DGCA)
library(proDA)
library(DESeq2)
library(cowplot)
library(igraph)
library(ggraph)
library(tidyverse)Prepare datasets
load("../output/proteomic_LUMOS_20200430.RData")
load("../../var/patmeta_200522.RData")
load("../../var/ddsrna_180717.RData")Preprocessing protein and RNA data
#subset samples and genes
overSampe <- intersect(colnames(dds), colnames(protCLL))
overGene <- intersect(rownames(dds), rowData(protCLL)$ensembl_gene_id)
ddsSub <- dds[overGene, overSampe]
protSub <- protCLL[match(overGene, rowData(protCLL)$ensembl_gene_id),overSampe]
rowData(ddsSub)$uniprotID <- rownames(protSub)[match(rownames(ddsSub),rowData(protSub)$ensembl_gene_id)]
#vst
ddsSub.vst <- varianceStabilizingTransformation(ddsSub)Processing protein complex data
int_pairs <- read_delim("../data/proteins_in_complexes", delim = "\t") %>%
mutate(Reactome = grepl("Reactome",Evidence_supporting_the_interaction),
Corum = grepl("Corum",Evidence_supporting_the_interaction)) %>%
filter(ProtA %in% rownames(protSub) & ProtB %in% rownames(protSub)) %>%
mutate(pair=map2_chr(ProtA, ProtB, ~paste0(sort(c(.x,.y)), collapse = "-"))) %>%
mutate(database = case_when(
Reactome & Corum ~ "both",
Reactome & !Corum ~ "Reactome",
!Reactome & Corum ~ "Corum",
TRUE ~ "other"
)) %>% mutate(inComplex = "yes")Construct protein-protein interaction network by “cause” proteins and “effect” proteins
fdrCut <- 0.1
resTab <- select(allRes, name, uniprotID, chrom, padj, padj.rna, logFC, log2FC.rna) %>%
mutate(sigProt = padj <= fdrCut,
sigRna = padj.rna <=fdrCut,
upProt = logFC > 0,
upRna = log2FC.rna >0)comTab <- int_pairs %>% select(ProtA, ProtB, database) %>%
left_join(resTab, by = c(ProtA = "uniprotID")) %>%
left_join(resTab, by = c(ProtB = "uniprotID"))
comTab.filter <- comTab %>%
filter(sigProt.x, sigProt.y, logFC.x*logFC.y >0) %>%
mutate(direct = ifelse(logFC.x >0, "stabilizing", "destabilizing")) %>%
mutate(source = case_when(
sigProt.x & sigRna.x & sigProt.y & !sigRna.y ~ name.x,
sigProt.y & sigRna.y & sigProt.x & !sigRna.x ~ name.y)) %>%
filter(!is.na(source)) %>%
mutate(target = ifelse(name.x == source, name.y, name.x)) %>%
select(source, target, direct)#get node list
allNodes <- union(comTab.filter$source, comTab.filter$target)
nodeList <- data.frame(id = seq(length(allNodes))-1, name = allNodes, stringsAsFactors = FALSE) %>%
mutate(causal = ifelse(name %in% comTab.filter$source, "cause", "effect"))
#get edge list
edgeList <- select(comTab.filter, source, target, direct) %>%
dplyr::rename(Source = source, Target = target) %>%
mutate(Source = nodeList[match(Source,nodeList$name),]$id,
Target = nodeList[match(Target, nodeList$name),]$id) %>%
data.frame(stringsAsFactors = FALSE)
net <- graph_from_data_frame(vertices = nodeList, d=edgeList, directed = FALSE)tidyNet <- as_tbl_graph(net)
ggraph(tidyNet) + geom_edge_link(aes(color = direct), width=1) +
geom_node_point(aes(color =causal, shape = causal), size=5) +
geom_node_text(aes(label = name), repel = TRUE) +
scale_color_manual(values = c(cause = "red",effect = "blue")) +
scale_edge_color_brewer(palette = "Set2") +
theme_graph() 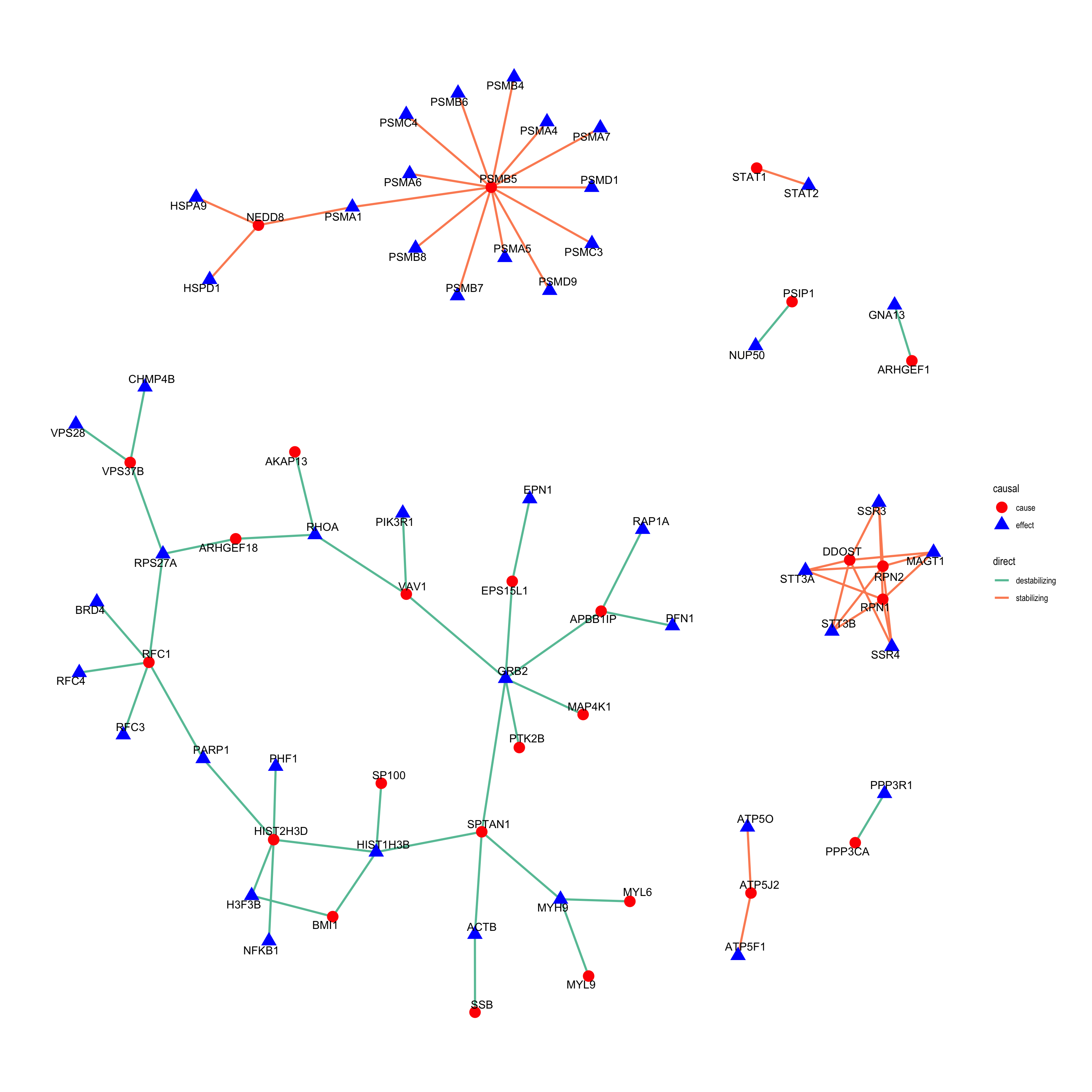 In this plot, the “cause” proteins, which show both RNA and protein changes, are shown as red circle and the “effect” proteins, which only show protein level changes are shown as blue triangles. Different to the trisomy12 analysis, the “cause” can be up-regulated or down-regulated. Therefore, I also defined a direction of the effect, which can be “stabilizing” or “destabilizing”. Stabilizing means the “cause” protein and “effect” protein are both up-regulated, indicating that the abundance of the “effect” protein is stabilized by forming complex with a protein up-regulated by U-IGHV. While “destabilizing” means both proteins are down-regulated, indicating the “effect” protein is destabilized by the down-regulation of another protein in the complexes due to U-IGHV.
In this plot, the “cause” proteins, which show both RNA and protein changes, are shown as red circle and the “effect” proteins, which only show protein level changes are shown as blue triangles. Different to the trisomy12 analysis, the “cause” can be up-regulated or down-regulated. Therefore, I also defined a direction of the effect, which can be “stabilizing” or “destabilizing”. Stabilizing means the “cause” protein and “effect” protein are both up-regulated, indicating that the abundance of the “effect” protein is stabilized by forming complex with a protein up-regulated by U-IGHV. While “destabilizing” means both proteins are down-regulated, indicating the “effect” protein is destabilized by the down-regulation of another protein in the complexes due to U-IGHV.
Compare to trisomy12, the network is much more sparse, which is reasonable as there are less proteins regulated by IGHV. But there are also some interesting pair. For example, GRB2, which are down-regulated in U-CLL at protein levels but not RNA levels, are connected to several proteins that are down-regulated at both RNA and protein levels in U-CLL. This can be explained as the down-regulations of those proteins leads to less protection effect to GRB2 through forming complexes in U-CLL and therefore, the protein abundance of GRB2 is decreased. On the other hand, the up-regulation of STAT1 on both RNA and protein levels seems to stabilize STAT2 in U-CLL through forming complex. Let me know if you spot other interesting cases that need further investigation.
Prepare complex table
edgeTab <- int_pairs %>% select(ProtA, ProtB, database) %>%
mutate(chrA = rowData(protCLL[ProtA,])$chromosome_name,
chrB = rowData(protCLL[ProtB,])$chromosome_name,
nameA = rowData(protCLL[ProtA,])$hgnc_symbol,
nameB = rowData(protCLL[ProtB,])$hgnc_symbol) %>%
filter(!is.na(chrA), !is.na(chrB)) %>%
select(ProtA, ProtB, nameA, nameB, database)fdrCut <- 0.1
nodeTab <- select(allRes, name, uniprotID, chrom, padj, padj.rna, logFC,log2FC.rna) %>%
mutate(sigProt = padj <= fdrCut,
sigRna = padj.rna <=fdrCut) %>%
mutate(changeRNA = case_when(
sigRna & log2FC.rna > 0 ~ "Up",
sigRna & log2FC.rna <0 ~ "Down",
TRUE ~ "n.s."
),
changeProtein = case_when(
sigProt & logFC > 0 ~ "Up",
sigProt & logFC < 0 ~ "Down",
TRUE ~ "n.s."
)) %>% select(name, chrom, changeRNA, changeProtein)STAT1-STAT2
plotSubNet <- function(protList,edgeTab, nodeTab) {
#get node list
subCom <- filter(edgeTab, nameA %in% protList | nameB %in% protList)
allNodes <- union(subCom$nameA, subCom$nameB)
nodeList <- data.frame(id = seq(length(allNodes))-1, name = allNodes, stringsAsFactors = FALSE) %>%
left_join(nodeTab, by = "name")
#get edge list
edgeList <- select(subCom, nameA, nameB, database) %>%
dplyr::rename(Source = nameA, Target = nameB) %>%
mutate(Source = nodeList[match(Source,nodeList$name),]$id,
Target = nodeList[match(Target, nodeList$name),]$id) %>%
data.frame(stringsAsFactors = FALSE)
net <- graph_from_data_frame(vertices = nodeList, d=edgeList, directed = FALSE)
tidyNet <- as_tbl_graph(net)
ggraph(tidyNet) + geom_edge_link(aes(color = database), width=1) +
geom_node_point(aes(color = changeProtein, shape = changeRNA), size=5) +
geom_node_text(aes(label = name), repel = TRUE) +
scale_color_manual(values = c(Up = "red", Down = "blue", n.s. = "grey")) +
scale_edge_color_brewer(palette = "Set2") +
theme_graph()
}protList <- c("STAT1","STAT2")
plotSubNet(protList, edgeTab, nodeTab)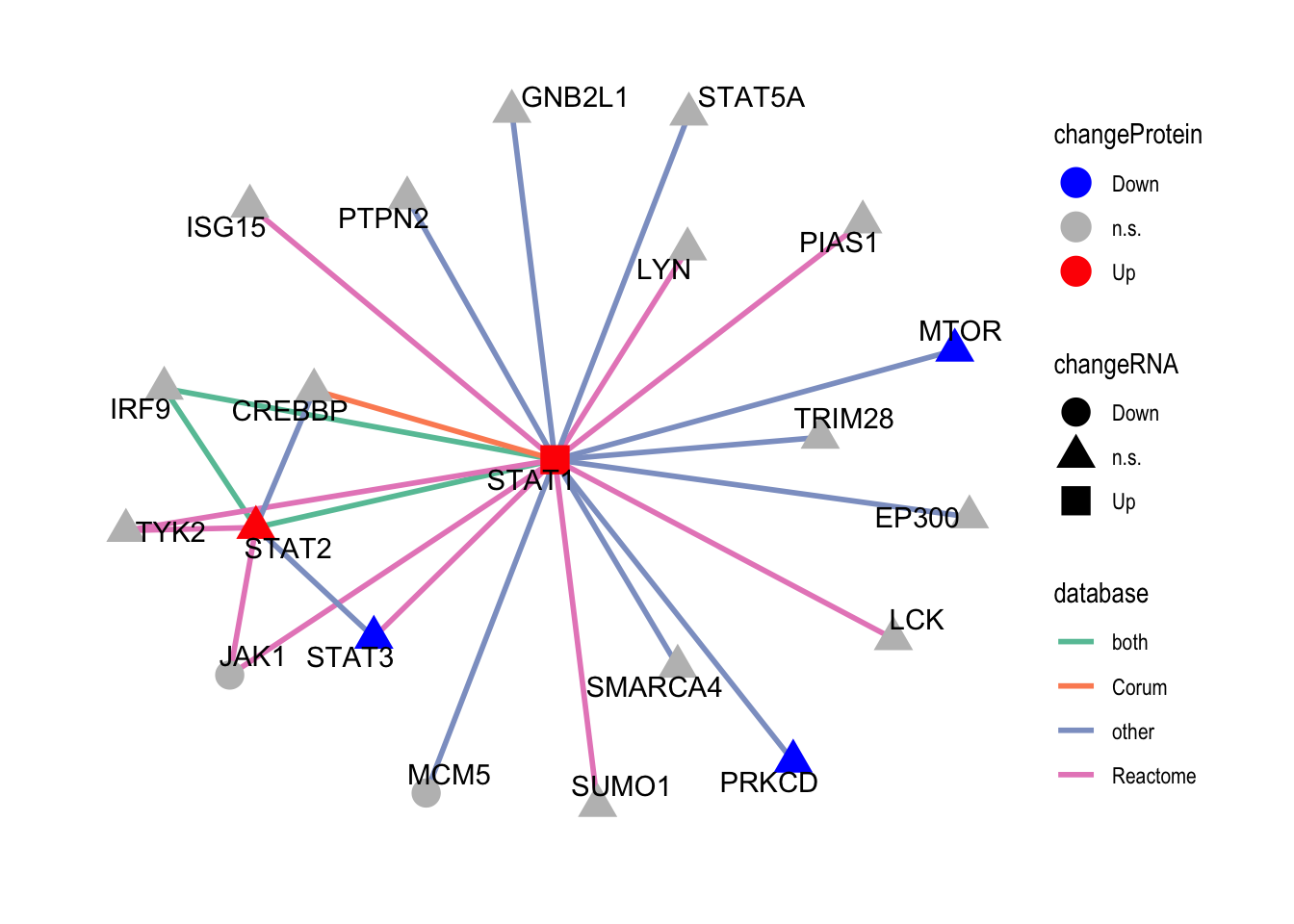
The color scheme here is a little different to the plot above or the trisomy12 network plot, because gene dosage effect is not involved and genes can be up-/down-regulated. In this plot, the color of the nodes indicates expression change at protein level, the shape of the nodes indicates expression change at RNA level.
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] forcats_0.4.0 stringr_1.4.0
[3] dplyr_0.8.5 purrr_0.3.3
[5] readr_1.3.1 tidyr_1.0.0
[7] tibble_3.0.0 tidyverse_1.3.0
[9] ggraph_1.0.2 igraph_1.2.4.1
[11] cowplot_0.9.4 ggplot2_3.3.0
[13] DESeq2_1.24.0 proDA_1.1.2
[15] DGCA_1.0.2 tidygraph_1.1.2
[17] SummarizedExperiment_1.14.0 DelayedArray_0.10.0
[19] BiocParallel_1.18.0 matrixStats_0.54.0
[21] Biobase_2.44.0 GenomicRanges_1.36.0
[23] GenomeInfoDb_1.20.0 IRanges_2.18.1
[25] S4Vectors_0.22.0 BiocGenerics_0.30.0
loaded via a namespace (and not attached):
[1] readxl_1.3.1 backports_1.1.4 Hmisc_4.2-0
[4] workflowr_1.6.0 plyr_1.8.4 splines_3.6.0
[7] robust_0.4-18.1 digest_0.6.19 foreach_1.4.4
[10] htmltools_0.4.0 viridis_0.5.1 GO.db_3.8.2
[13] magrittr_1.5 checkmate_2.0.0 memoise_1.1.0
[16] fit.models_0.5-14 cluster_2.1.0 doParallel_1.0.14
[19] fastcluster_1.1.25 annotate_1.62.0 modelr_0.1.5
[22] colorspace_1.4-1 rvest_0.3.5 blob_1.1.1
[25] rrcov_1.4-9 ggrepel_0.8.1 haven_2.2.0
[28] xfun_0.8 crayon_1.3.4 RCurl_1.95-4.12
[31] jsonlite_1.6 genefilter_1.66.0 impute_1.58.0
[34] survival_2.44-1.1 iterators_1.0.10 glue_1.3.2
[37] polyclip_1.10-0 gtable_0.3.0 zlibbioc_1.30.0
[40] XVector_0.24.0 DEoptimR_1.0-8 scales_1.1.0
[43] mvtnorm_1.0-11 DBI_1.0.0 Rcpp_1.0.1
[46] viridisLite_0.3.0 xtable_1.8-4 htmlTable_1.13.1
[49] foreign_0.8-71 bit_1.1-14 preprocessCore_1.46.0
[52] Formula_1.2-3 htmlwidgets_1.3 httr_1.4.1
[55] RColorBrewer_1.1-2 acepack_1.4.1 ellipsis_0.2.0
[58] pkgconfig_2.0.2 XML_3.98-1.20 farver_2.0.3
[61] nnet_7.3-12 dbplyr_1.4.2 locfit_1.5-9.1
[64] dynamicTreeCut_1.63-1 labeling_0.3 tidyselect_1.0.0
[67] rlang_0.4.5 later_0.8.0 AnnotationDbi_1.46.0
[70] cellranger_1.1.0 munsell_0.5.0 tools_3.6.0
[73] cli_1.1.0 generics_0.0.2 RSQLite_2.1.1
[76] broom_0.5.2 evaluate_0.14 yaml_2.2.0
[79] knitr_1.23 bit64_0.9-7 fs_1.4.0
[82] robustbase_0.93-5 nlme_3.1-140 xml2_1.2.2
[85] compiler_3.6.0 rstudioapi_0.10 reprex_0.3.0
[88] tweenr_1.0.1 geneplotter_1.62.0 pcaPP_1.9-73
[91] stringi_1.4.3 lattice_0.20-38 Matrix_1.2-17
[94] vctrs_0.2.4 pillar_1.4.3 lifecycle_0.2.0
[97] data.table_1.12.2 bitops_1.0-6 httpuv_1.5.1
[100] R6_2.4.0 latticeExtra_0.6-28 promises_1.0.1
[103] gridExtra_2.3 codetools_0.2-16 MASS_7.3-51.4
[106] assertthat_0.2.1 rprojroot_1.3-2 withr_2.1.2
[109] GenomeInfoDbData_1.2.1 hms_0.5.2 grid_3.6.0
[112] rpart_4.1-15 rmarkdown_1.13 git2r_0.26.1
[115] ggforce_0.2.2 lubridate_1.7.4 WGCNA_1.68
[118] base64enc_0.1-3