Explaining STAT2 protein expression using multipmics data
Junyan Lu
2020-03-09
Last updated: 2020-09-14
Checks: 5 2
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you'll want to first commit it to the Git repo. If you're still working on the analysis, you can ignore this warning. When you're finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- unnamed-chunk-12
- unnamed-chunk-25
- unnamed-chunk-28
- unnamed-chunk-4
- unnamed-chunk-5
To ensure reproducibility of the results, delete the cache directory lassoForSTAT2_cache and re-run the analysis. To have workflowr automatically delete the cache directory prior to building the file, set delete_cache = TRUE when running wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3fb50c5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/correlateCLLPD_cache/
Ignored: analysis/lassoForSTAT2_cache/
Ignored: analysis/manuscript_S1_Overview_cache/
Ignored: analysis/manuscript_S2_genomicAssociation_oldTimsTOF_cache/
Ignored: analysis/manuscript_S4_IGHV_oldTimsTOF_cache/
Ignored: analysis/manuscript_S6_del11q_oldTimsTOF_cache/
Ignored: analysis/manuscript_S8_drugResponse_Outcomes_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/.trisomy12_norm.pdf
Untracked: analysis/CNVanalysis_11q.Rmd
Untracked: analysis/CNVanalysis_trisomy12.Rmd
Untracked: analysis/CNVanalysis_trisomy19.Rmd
Untracked: analysis/STAT2_cytokines.Rmd
Untracked: analysis/SUGP1_splicing.svg.xml
Untracked: analysis/analysisDrugResponses.Rmd
Untracked: analysis/analysisDrugResponses_IC50.Rmd
Untracked: analysis/analysisPCA.Rmd
Untracked: analysis/analysisPreliminary_LUMOS.Rmd
Untracked: analysis/analysisPreliminary_timsTOF_Hela.Rmd
Untracked: analysis/analysisPreliminary_timsTOF_new.Rmd
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/comparePlatforms_LUMOS_helaTimsTOF.Rmd
Untracked: analysis/comparePlatforms_LUMOS_newTimsTOF.Rmd
Untracked: analysis/comparePlatforms_newTimsTOF_helaTimsTOF.Rmd
Untracked: analysis/complexAnalysis_IGHV.Rmd
Untracked: analysis/complexAnalysis_IGHV_alternative.Rmd
Untracked: analysis/complexAnalysis_overall.Rmd
Untracked: analysis/complexAnalysis_trisomy12.Rmd
Untracked: analysis/complexAnalysis_trisomy12_alternative.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/correlateGenomic_timsTOFnew.Rmd
Untracked: analysis/correlateGenomic_timsTOFnewHela.Rmd
Untracked: analysis/correlateRNAexpression.Rmd
Untracked: analysis/default.css
Untracked: analysis/del11q.pdf
Untracked: analysis/del11q_norm.pdf
Untracked: analysis/full_diff_list.csv
Untracked: analysis/lassoForSTAT2.Rmd
Untracked: analysis/manuscript_S0_PrepareData.Rmd
Untracked: analysis/manuscript_S1_Overview.Rmd
Untracked: analysis/manuscript_S2_genomicAssociation.Rmd
Untracked: analysis/manuscript_S2_genomicAssociation_oldTimsTOF.Rmd
Untracked: analysis/manuscript_S3_trisomy12.Rmd
Untracked: analysis/manuscript_S4_IGHV.Rmd
Untracked: analysis/manuscript_S4_IGHV_oldTimsTOF.Rmd
Untracked: analysis/manuscript_S5_trisomy19.Rmd
Untracked: analysis/manuscript_S6_del11q.Rmd
Untracked: analysis/manuscript_S6_del11q_oldTimsTOF.Rmd
Untracked: analysis/manuscript_S7_SF3B1.Rmd
Untracked: analysis/manuscript_S8_drugResponse_Outcomes.Rmd
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/plotCNV_del11q.pdf
Untracked: analysis/plotExpressionCNV.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/processProteomics_timsTOF_Hela.Rmd
Untracked: analysis/processProteomics_timsTOF_new.Rmd
Untracked: analysis/protCLL.RData
Untracked: analysis/qualityControl_timsTOF_Hela.Rmd
Untracked: analysis/qualityControl_timsTOF_new.Rmd
Untracked: analysis/style.css
Untracked: analysis/tableS1_DE_proteins_p0.01.xlsx
Untracked: analysis/test.pdf
Untracked: analysis/test.svg
Untracked: analysis/tri12Enrich.pdf
Untracked: analysis/trisomy12.pdf
Untracked: analysis/trisomy12_AFcor.Rmd
Untracked: analysis/trisomy12_norm.pdf
Untracked: code/AlteredPQR.R
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/200725_cll_diaPASEF_direct_reports/
Untracked: data/200728_cll_diaPASEF_direct_plus_hela_reports/
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/Chemokines.csv
Untracked: data/IFN_list.csv
Untracked: data/IFNreceptor.csv
Untracked: data/Interleukin_receptor.csv
Untracked: data/Interleukins.csv
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/chemoReceptor.csv
Untracked: data/example_proteomics_data
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/proteins_in_complexes
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/Fig1A.pdf
Untracked: output/Fig1A.png
Untracked: output/Fig1A.pptx
Untracked: output/LUMOS_processed.RData
Untracked: output/MSH6_splicing.svg
Untracked: output/SUGP1_splicing.eps
Untracked: output/SUGP1_splicing.pdf
Untracked: output/SUGP1_splicing.svg
Untracked: output/cnv_plots.zip
Untracked: output/cnv_plots/
Untracked: output/cnv_plots_norm.zip
Untracked: output/ddsrna_enc.RData
Untracked: output/deResList.RData
Untracked: output/deResList_timsTOF.RData
Untracked: output/deResList_timsTOF_old.RData
Untracked: output/dxdCLL.RData
Untracked: output/dxdCLL2.RData
Untracked: output/encMap.RData
Untracked: output/exprCNV.RData
Untracked: output/exprCNV_enc.RData
Untracked: output/lassoResults_CPS.RData
Untracked: output/lassoResults_IC50.RData
Untracked: output/patMeta_enc.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepCLL_lumos_enc.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/pheno1000_enc.RData
Untracked: output/pheno1000_main.RData
Untracked: output/plotCNV_allChr11_diff.pdf
Untracked: output/plotCNV_del11q_sum.pdf
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_LUMOS_20200430.RData
Untracked: output/proteomic_LUMOS_enc.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/proteomic_timsTOF_Hela_20200806.RData
Untracked: output/proteomic_timsTOF_enc.RData
Untracked: output/proteomic_timsTOF_new_20200806.RData
Untracked: output/proteomic_timsTOF_old_enc.RData
Untracked: output/splicingResults.RData
Untracked: output/survival_enc.RData
Untracked: output/timsTOF_processed.RData
Untracked: plotCNV_del11q_diff.pdf
Untracked: summary/
Untracked: supp_latex/
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateCLLPD.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Feature selection with LASSO
Preprocessing data
Proteomics data
[1] 49 576RNAseq
#subset
ddsSub <- dds[,dds$PatID %in% colnames(protCLL)]
#only keep protein coding genes with symbol
ddsSub <- ddsSub[rowData(ddsSub)$biotype %in% "protein_coding" & !rowData(ddsSub)$symbol %in% c("",NA),]
#remove lowly expressed genes
ddsSub <- ddsSub[rowSums(counts(ddsSub, normalized = TRUE)) > 100,]
#voom transformation
exprMat <- limma::voom(counts(ddsSub), lib.size = ddsSub$sizeFactor)$E
ddsSub.voom <- ddsSub
assay(ddsSub.voom) <- exprMat
rnaMat <- exprMat
rownames(rnaMat) <- rowData(ddsSub.voom)$symbol
# Prefiltering
overSampe <- intersect(names(yVec), colnames(rnaMat))
designMat <- model.matrix(~ yVec[overSampe])
fit <- lmFit(rnaMat[,overSampe], design = designMat)
fit2 <- eBayes(fit)
resTab <- topTable(fit2, number = Inf) %>% data.frame() %>% rownames_to_column("ID")
keepRna <- filter(resTab, adj.P.Val < 0.05)$ID
rnaMat <- t(rnaMat[keepRna, ])
dim(rnaMat)[1] 46 792colnames(rnaMat) <- paste0(colnames(rnaMat),".rna")Genomic data
ighvMap <- c(M = 1, U=0)
methMap <- c(LP= 0, IP=0.5, HP=1 )
#genetics
genData <- filter(patMeta, Patient.ID %in% colnames(protCLL)) %>%
select(-HIPO.ID, -project, -date.of.diagnosis, -treatment, -date.of.first.treatment,
-gender, -diagnosis) %>%
dplyr::rename(IGHV = IGHV.status, MClust= Methylation_Cluster) %>%
mutate_at(vars(-Patient.ID), as.character) %>%
mutate(IGHV = ighvMap[IGHV], MClust = methMap[MClust]) %>%
mutate_at(vars(-Patient.ID), as.numeric) %>%
data.frame() %>% column_to_rownames("Patient.ID")
#remove gene with higher than 40% missing values
genData <- genData[,colSums(is.na(genData))/nrow(genData) <= 0.4]
#remove genes with less than 5 mutated cases
genData <- genData[,colSums(genData, na.rm = TRUE) >= 5]
#fill the missing value with majority
genData <- apply(genData, 2, function(x) {
xVec <- x
avgVal <- mean(x,na.rm= TRUE)
if (avgVal >= 0.5) {
xVec[is.na(xVec)] <- 1
} else xVec[is.na(xVec)] <- 0
xVec
})Drug responses
#choose the first sample
viabMat <- arrange(pheno1000_main, screenDate) %>%
filter(diagnosis == "CLL", patientID %in% colnames(protCLL)) %>%
distinct(patientID, Drug, concIndex, .keep_all = TRUE) %>%
filter(! Drug %in% c("DMSO","PBS")) %>%
mutate(id = paste0(Drug,"_",concIndex)) %>%
select(patientID, id, normVal.adj.sigm) %>%
spread(key = patientID, value = normVal.adj.sigm) %>%
data.frame() %>% column_to_rownames("id") %>%
as.matrix() %>% t()Feature selection with LASSO penalty
#Functions for running glm
runGlm <- function(X, y, method = "ridge", repeats=20, folds = 3, lambda = "lambda.1se") {
modelList <- list()
lambdaList <- c()
varExplain <- c()
coefMat <- matrix(NA, ncol(X), repeats)
rownames(coefMat) <- colnames(X)
if (method == "lasso"){
alpha = 1
} else if (method == "ridge") {
alpha = 0
}
for (i in seq(repeats)) {
if (ncol(X) > 2) {
res <- cv.glmnet(X,y, type.measure = "mse", family="gaussian",
nfolds = folds, alpha = alpha, standardize = FALSE)
lambdaList <- c(lambdaList, res[[lambda]])
modelList[[i]] <- res
coefModel <- coef(res, s = lambda)[-1] #remove intercept row
coefMat[,i] <- coefModel
#calculate variance explained
y.pred <- predict(res, s = lambda, newx = X)
varExp <- cor(as.vector(y),as.vector(y.pred))^2
varExplain[i] <- ifelse(is.na(varExp), 0, varExp)
} else {
fitlm<-lm(y~., data.frame(X))
varExp <- summary(fitlm)$r.squared
varExplain <- c(varExplain, varExp)
}
}
list(modelList = modelList, lambdaList = lambdaList, varExplain = varExplain, coefMat = coefMat)
}#function for scaling predictors
dataScale <- function(x, censor = NULL, robust = FALSE) {
#function to scale different variables
if (length(unique(na.omit(x))) <=3){
#a binary variable, change to -0.5 and 0.5 for 1 and 2
x - 0.5
} else {
if (robust) {
#continuous variable, centered by median and divied by 2*mad
mScore <- (x-median(x,na.rm=TRUE))/mad(x,na.rm=TRUE)
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
} else {
mScore <- (x-mean(x,na.rm=TRUE))/(sd(x,na.rm=TRUE))
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
}
}
}Clean and integrate multi-omics data
Variance explained for STAT2 expression by multi-omics datasets
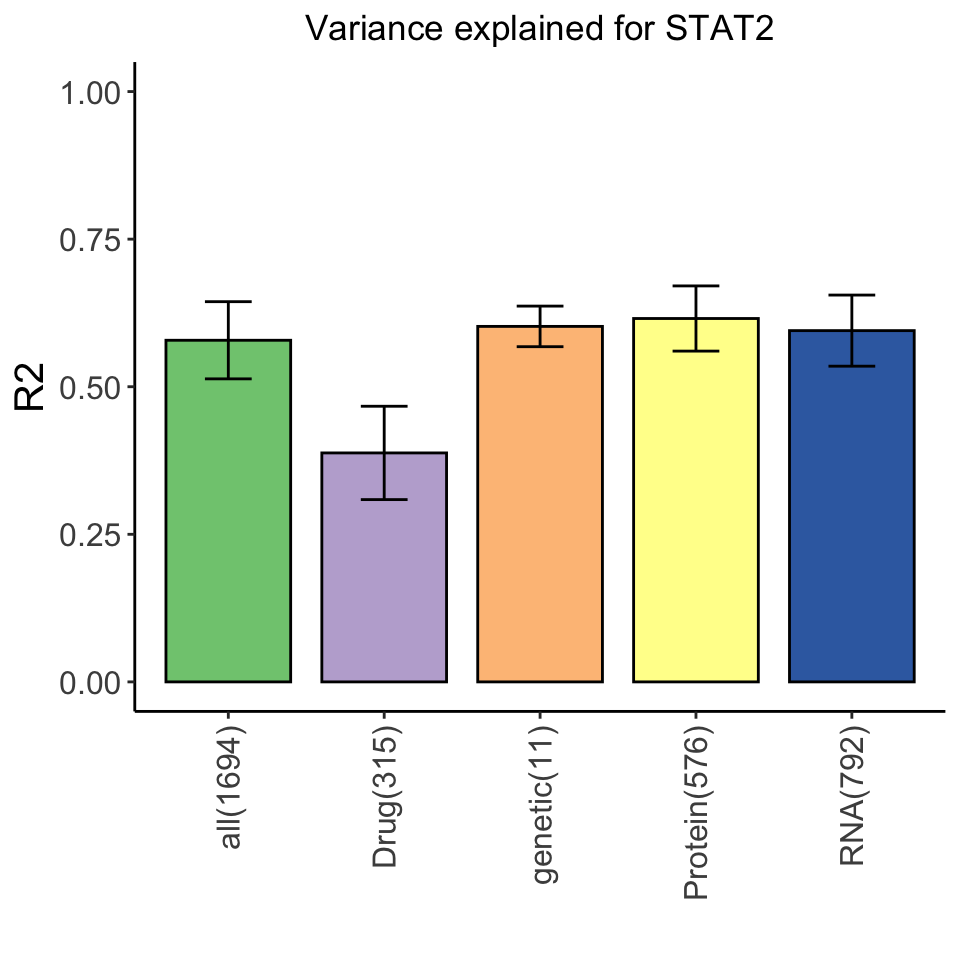 Based on this plot, genetics alone alreay explains STAT2 expression quite well. Other datasets do not add much information
Based on this plot, genetics alone alreay explains STAT2 expression quite well. Other datasets do not add much information
Heatmap of selected features
Genetics only

STAT2 protein expression stratified by IGHV and trisomy12
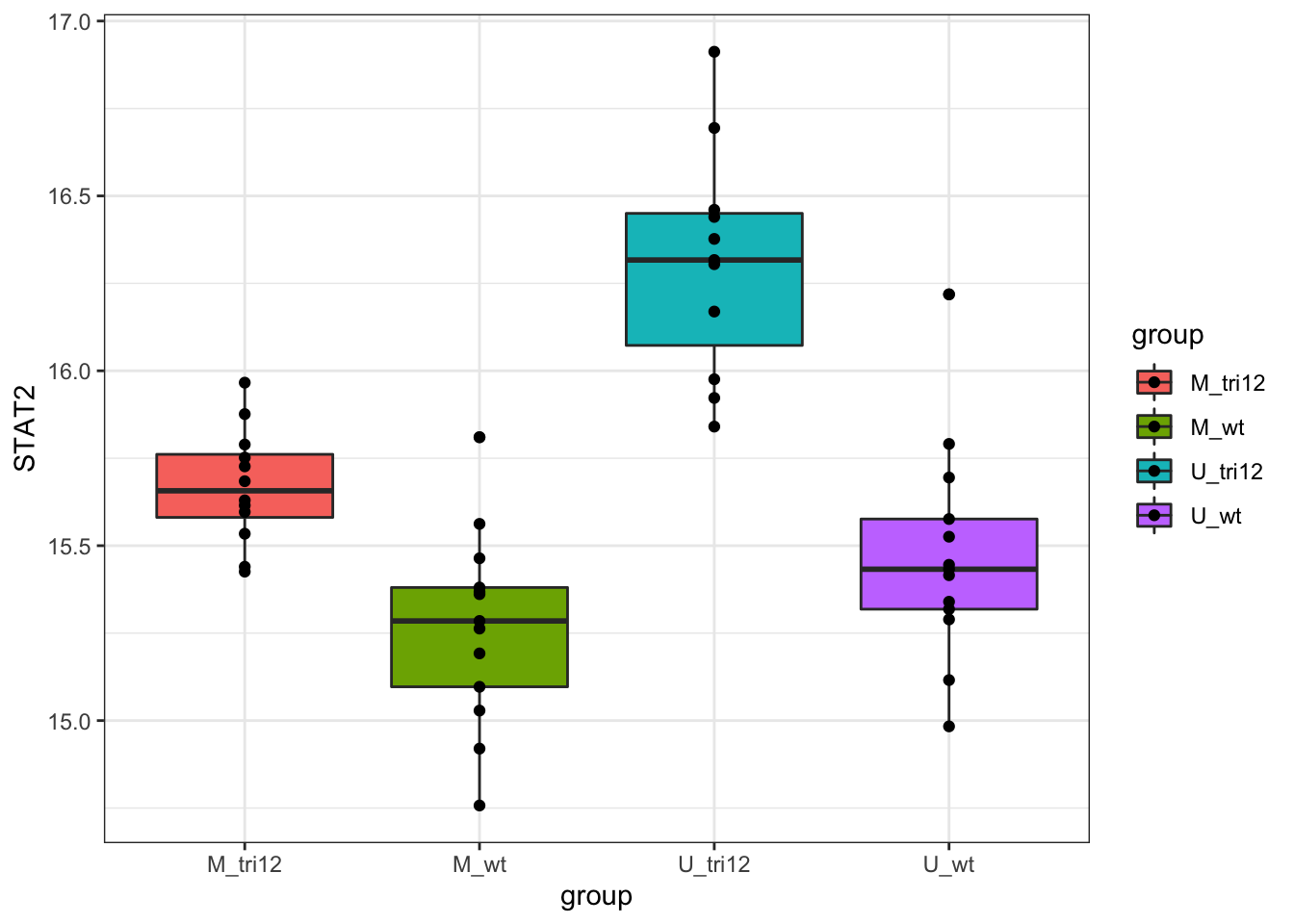
Call:
lm(formula = STAT2 ~ IGHV * trisomy12, data = plotTab)
Residuals:
Min 1Q Median 3Q Max
-0.51109 -0.15429 -0.00532 0.11960 0.74576
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 15.66963 0.07988 196.174 < 2e-16 ***
IGHVU 0.64067 0.11550 5.547 1.46e-06 ***
trisomy12wt -0.40129 0.11077 -3.623 0.000738 ***
IGHVU:trisomy12wt -0.43619 0.15849 -2.752 0.008504 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.2767 on 45 degrees of freedom
Multiple R-squared: 0.6733, Adjusted R-squared: 0.6515
F-statistic: 30.92 on 3 and 45 DF, p-value: 5.271e-11STAT2 protein expression is strongly affected by IGHV and trisomy12 status, U-CLLs with trisomy12 shows significant up-regulation of STAT2
STAT2 RNA expression stratified by IGHV and trisomy12
Samples in the proteomic cohort
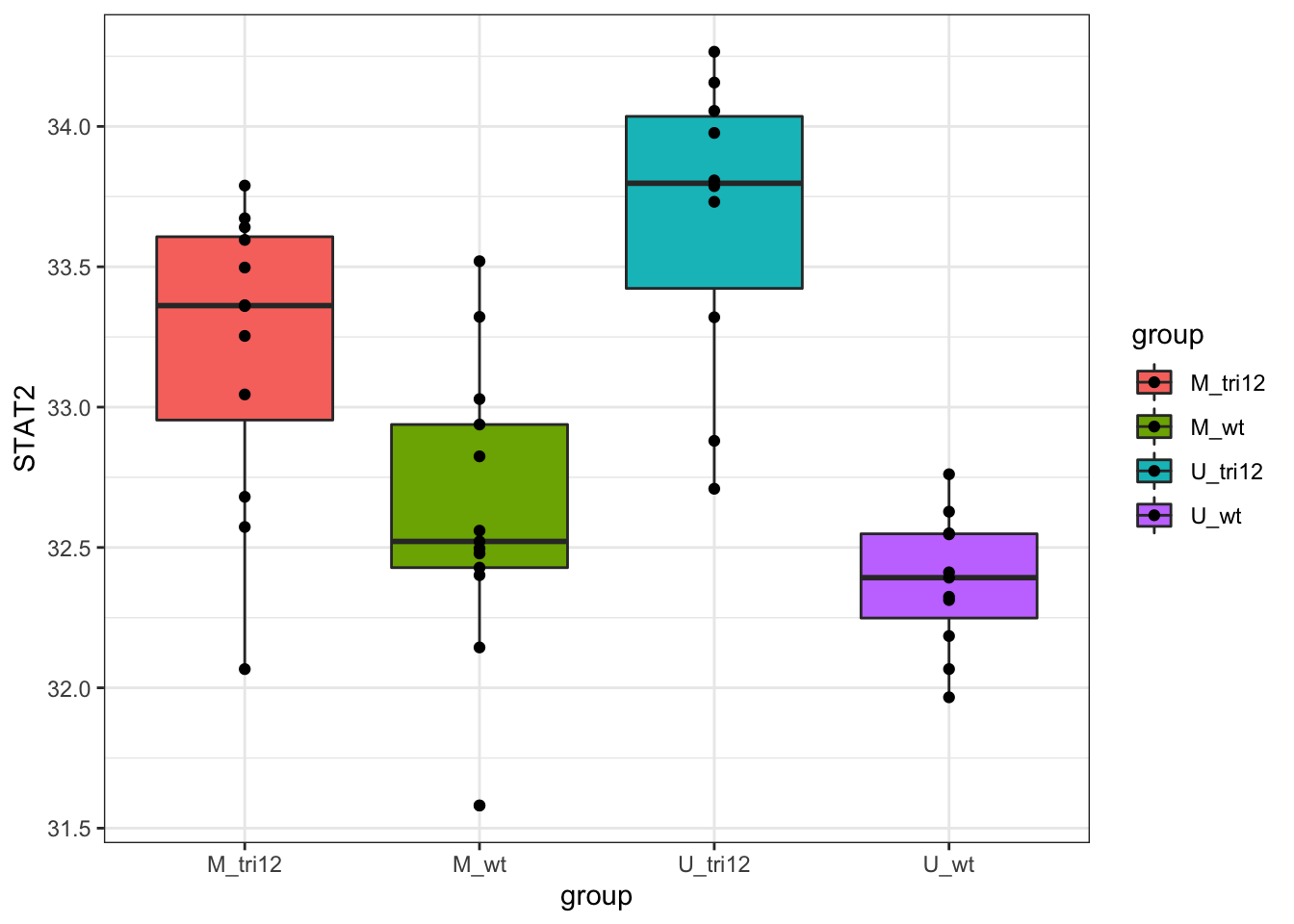
Call:
lm(formula = STAT2 ~ IGHV * trisomy12, data = plotTab)
Residuals:
Min 1Q Median 3Q Max
-1.14476 -0.20254 0.05228 0.30685 0.88566
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 33.2115 0.1351 245.761 < 2e-16 ***
IGHVU 0.4575 0.2004 2.283 0.02758 *
trisomy12wt -0.5774 0.1874 -3.081 0.00363 **
IGHVU:trisomy12wt -0.7149 0.2774 -2.577 0.01357 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.4681 on 42 degrees of freedom
Multiple R-squared: 0.5422, Adjusted R-squared: 0.5095
F-statistic: 16.58 on 3 and 42 DF, p-value: 2.951e-07Similar trend can be oberserved in RNAseq data, although not as significant as protein expression
Samples in the whole CLL RNAseq cohort
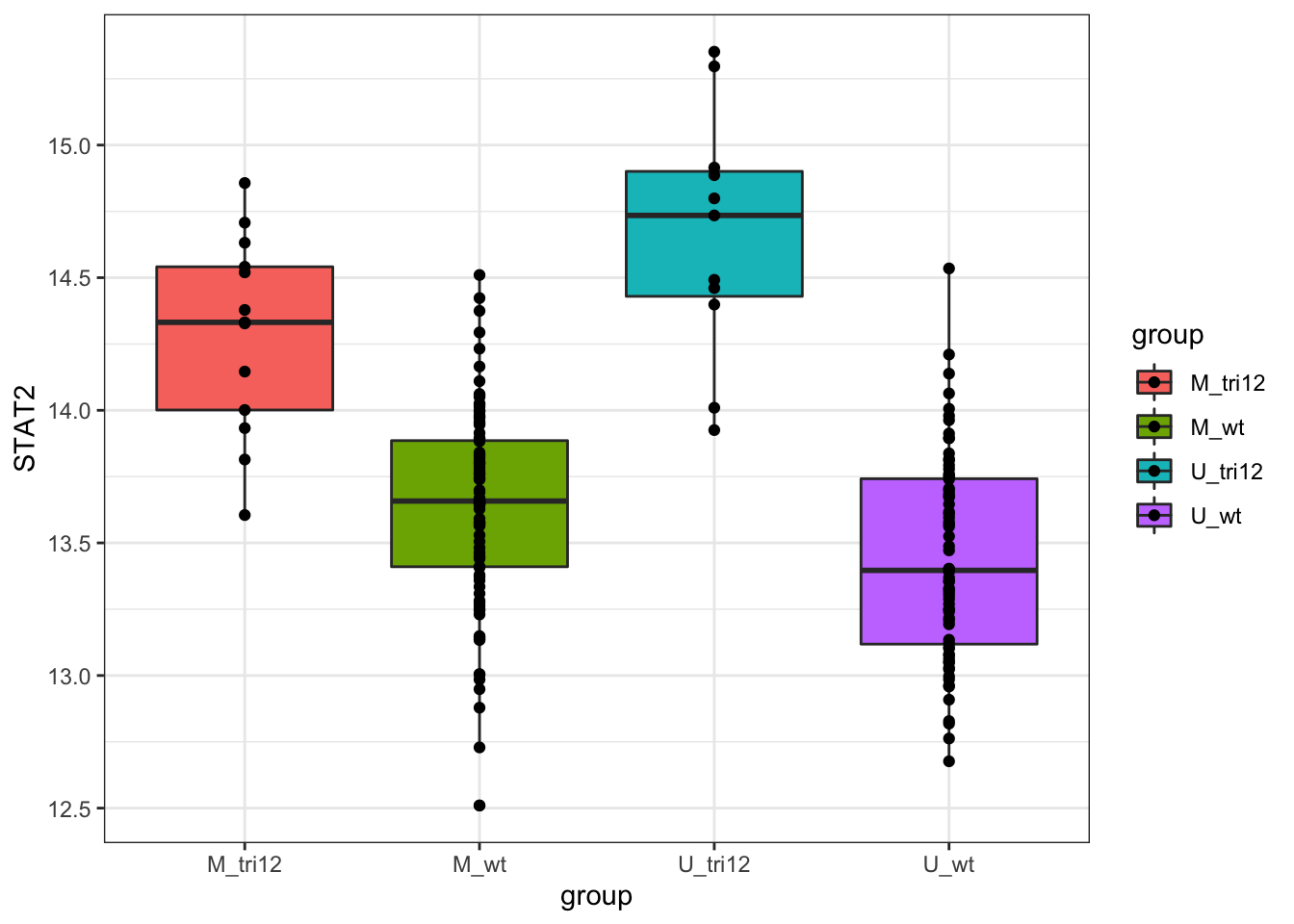
Call:
lm(formula = STAT2 ~ IGHV * trisomy12, data = plotTab)
Residuals:
Min 1Q Median 3Q Max
-1.12608 -0.26268 0.02179 0.25784 1.09138
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 14.2919 0.1057 135.197 < 2e-16 ***
IGHVU 0.3691 0.1561 2.364 0.019117 *
trisomy12wt -0.6559 0.1126 -5.823 2.47e-08 ***
IGHVU:trisomy12wt -0.5616 0.1671 -3.362 0.000939 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.3811 on 188 degrees of freedom
Multiple R-squared: 0.4182, Adjusted R-squared: 0.4089
F-statistic: 45.05 on 3 and 188 DF, p-value: < 2.2e-16RNA only
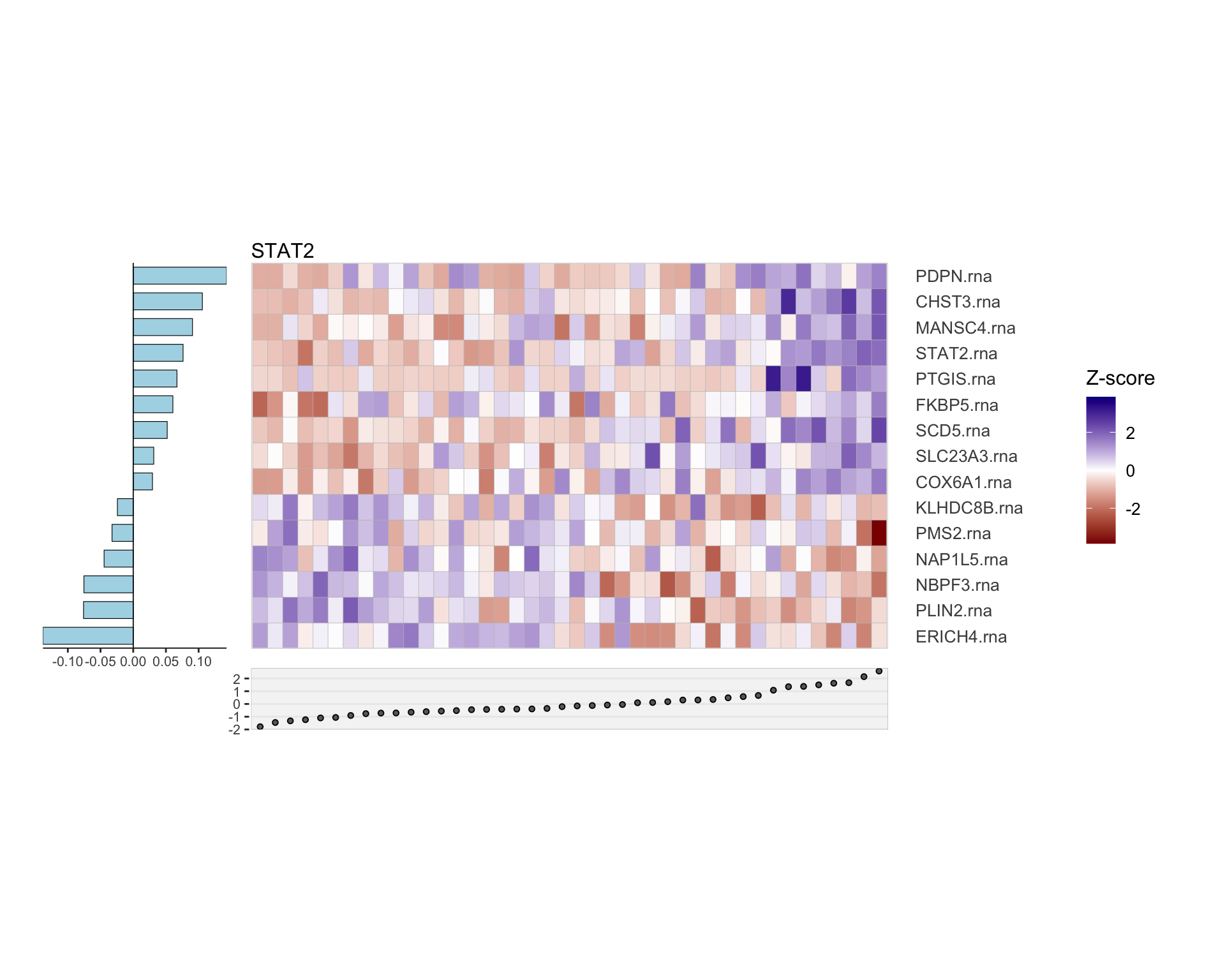
Protein only
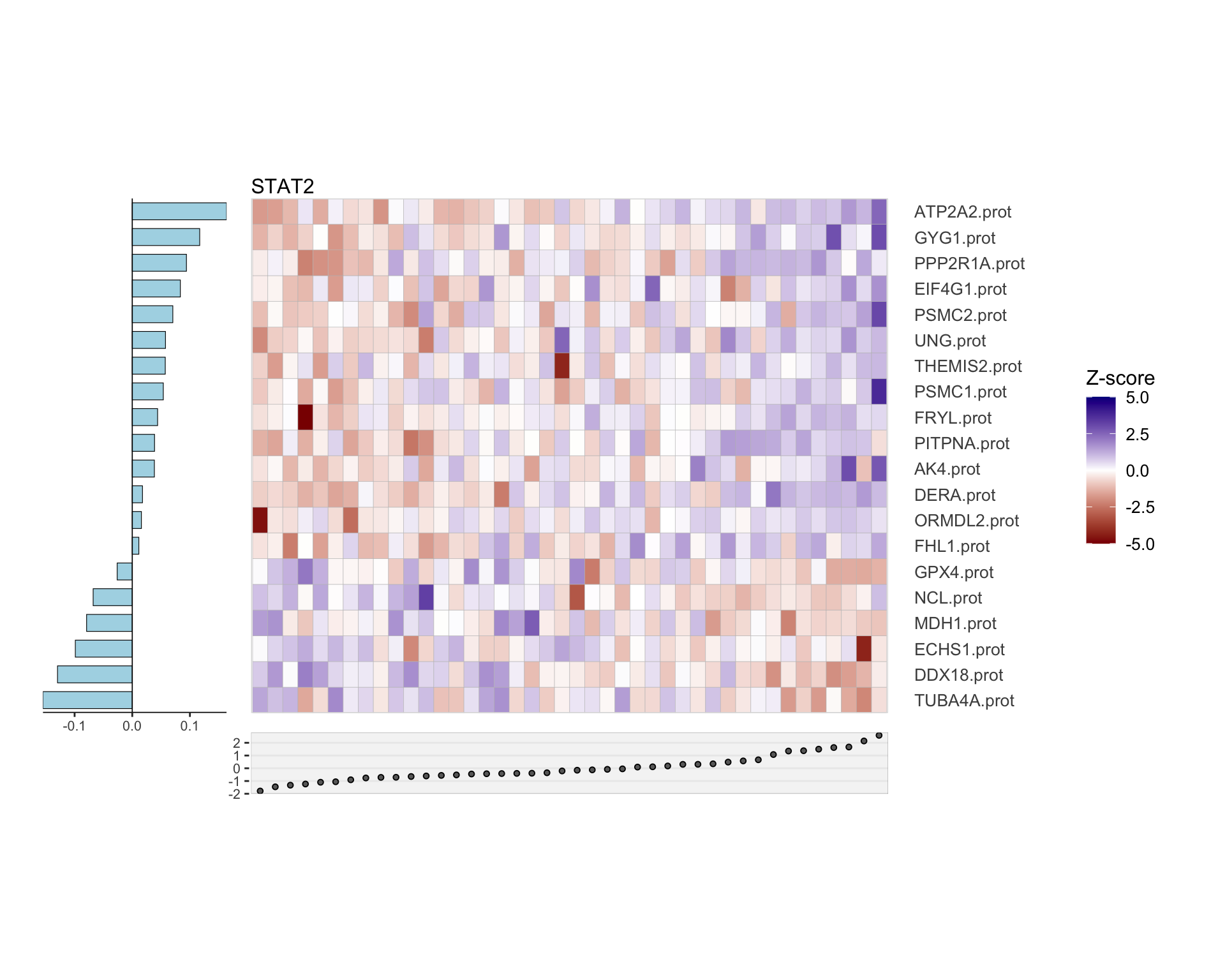
Drug responses only
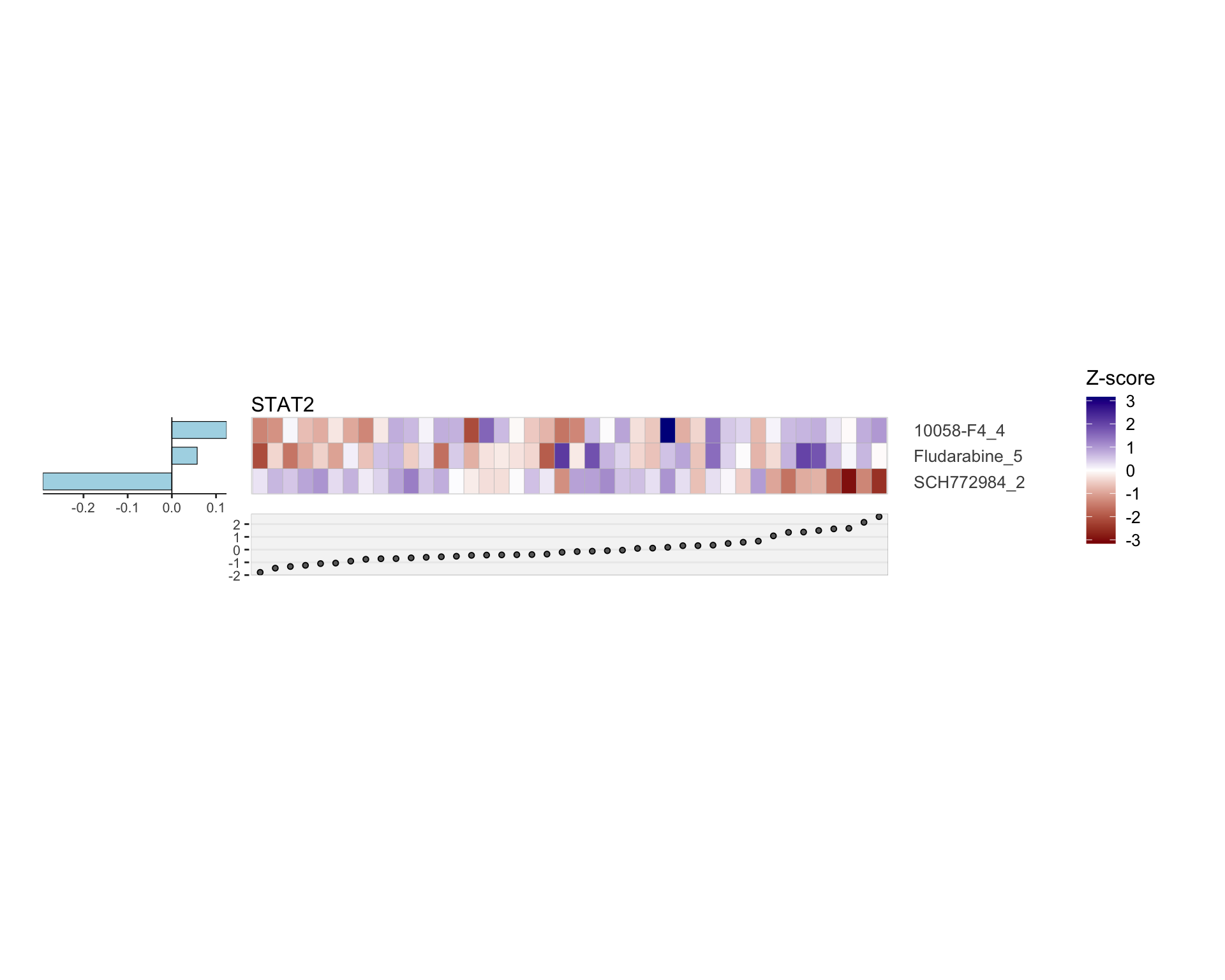
Combined
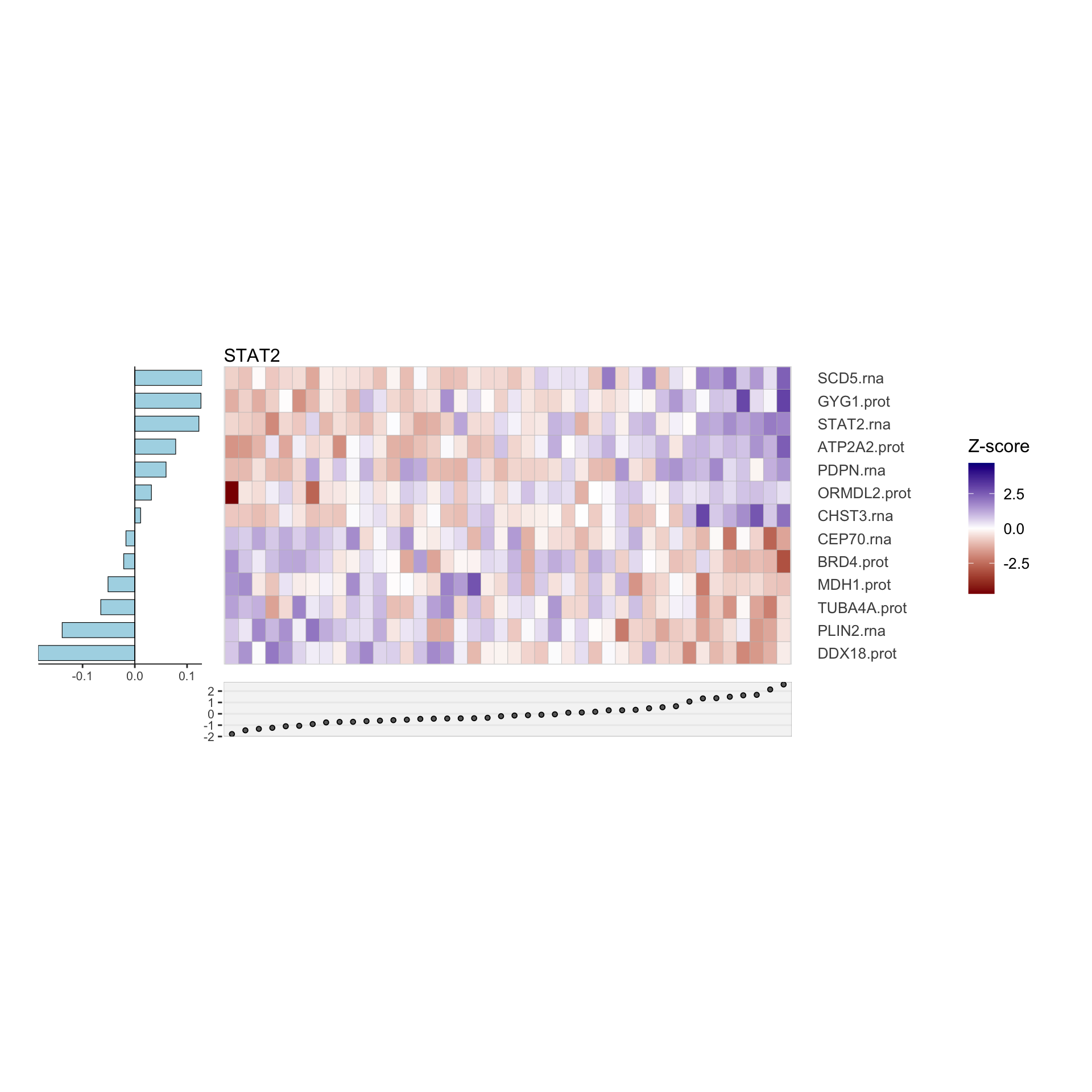
Plot all heatmaps
Pathway enrichment for RNA expressions correlated with STAT2 protein expression
Prepare data
#subset
ddsSub <- dds[,dds$PatID %in% names(yVec)]
#only keep protein coding genes with symbol
ddsSub <- ddsSub[rowData(ddsSub)$biotype %in% "protein_coding" & !rowData(ddsSub)$symbol %in% c("",NA),]
#remove lowly expressed genes
ddsSub <- ddsSub[rowSums(counts(ddsSub, normalized = TRUE)) > 100,]
#voom transformation
exprMat <- limma::voom(counts(ddsSub), lib.size = ddsSub$sizeFactor)$E
ddsSub.voom <- ddsSub
assay(ddsSub.voom) <- exprMat
rnaMat <- exprMat
rownames(rnaMat) <- rowData(ddsSub.voom)$symbol
overSampe <- intersect(names(yVec), colnames(rnaMat))
rnaMat <- rnaMat[,overSampe]
yVec <- yVec[overSampe]Test
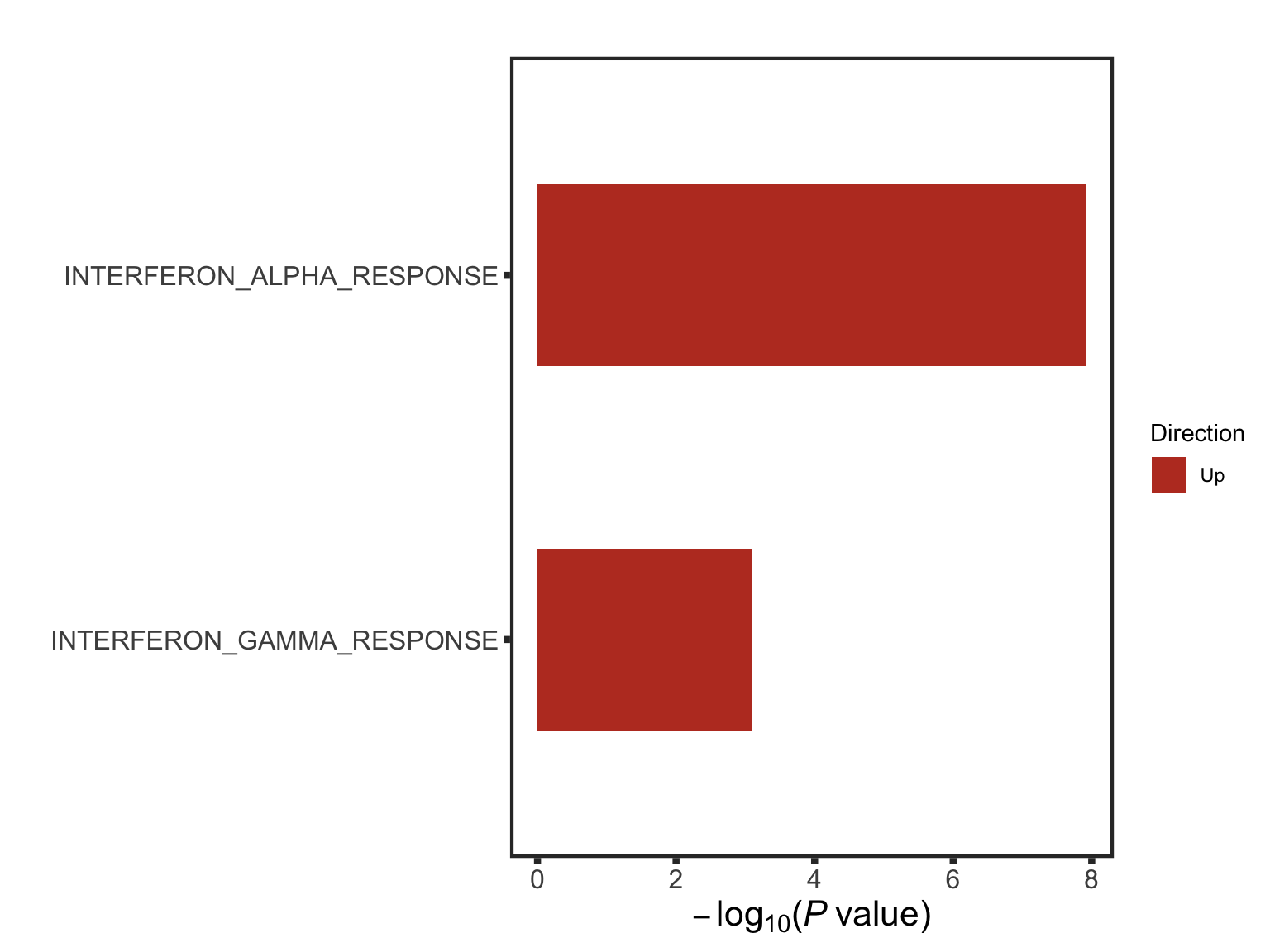
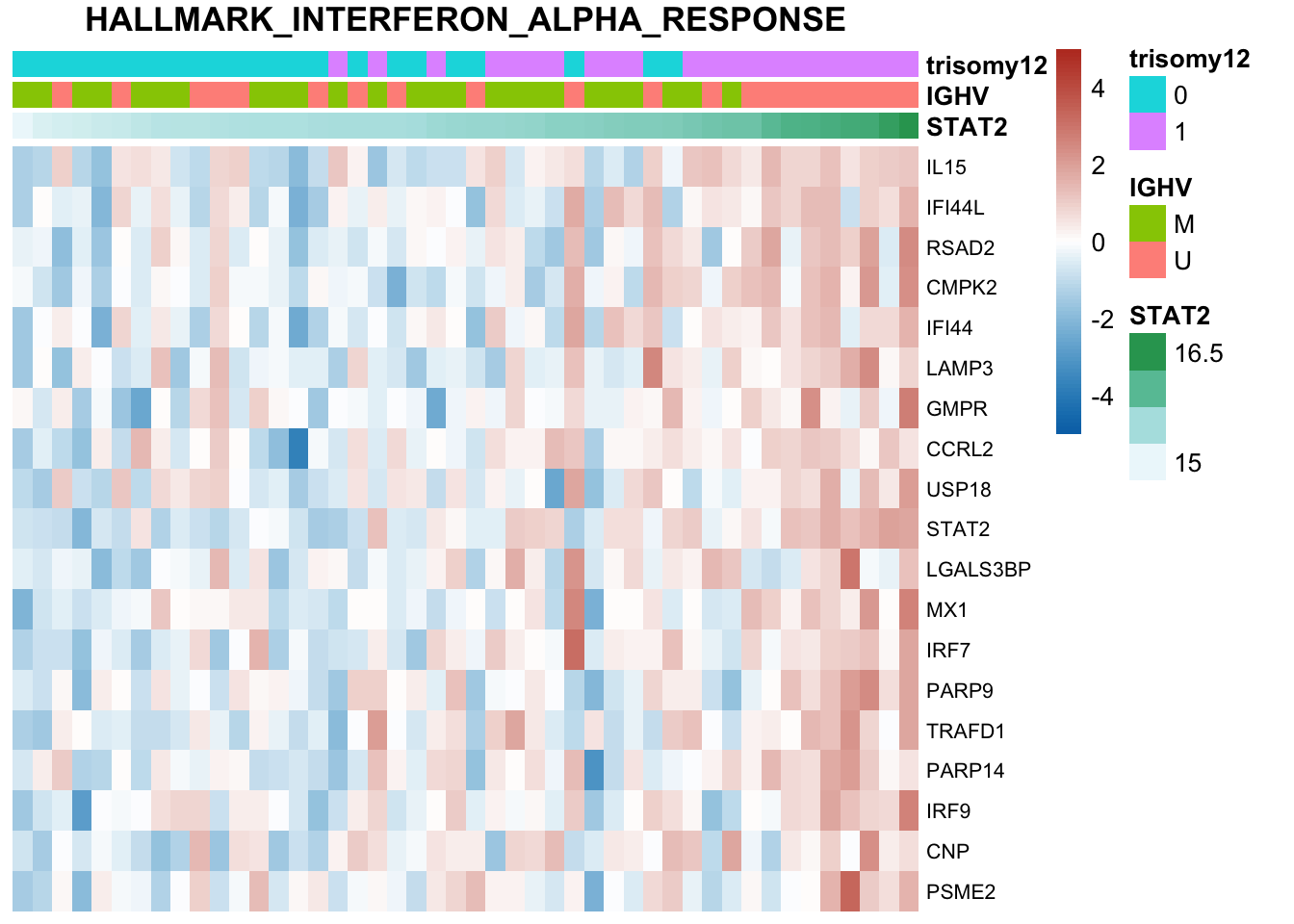
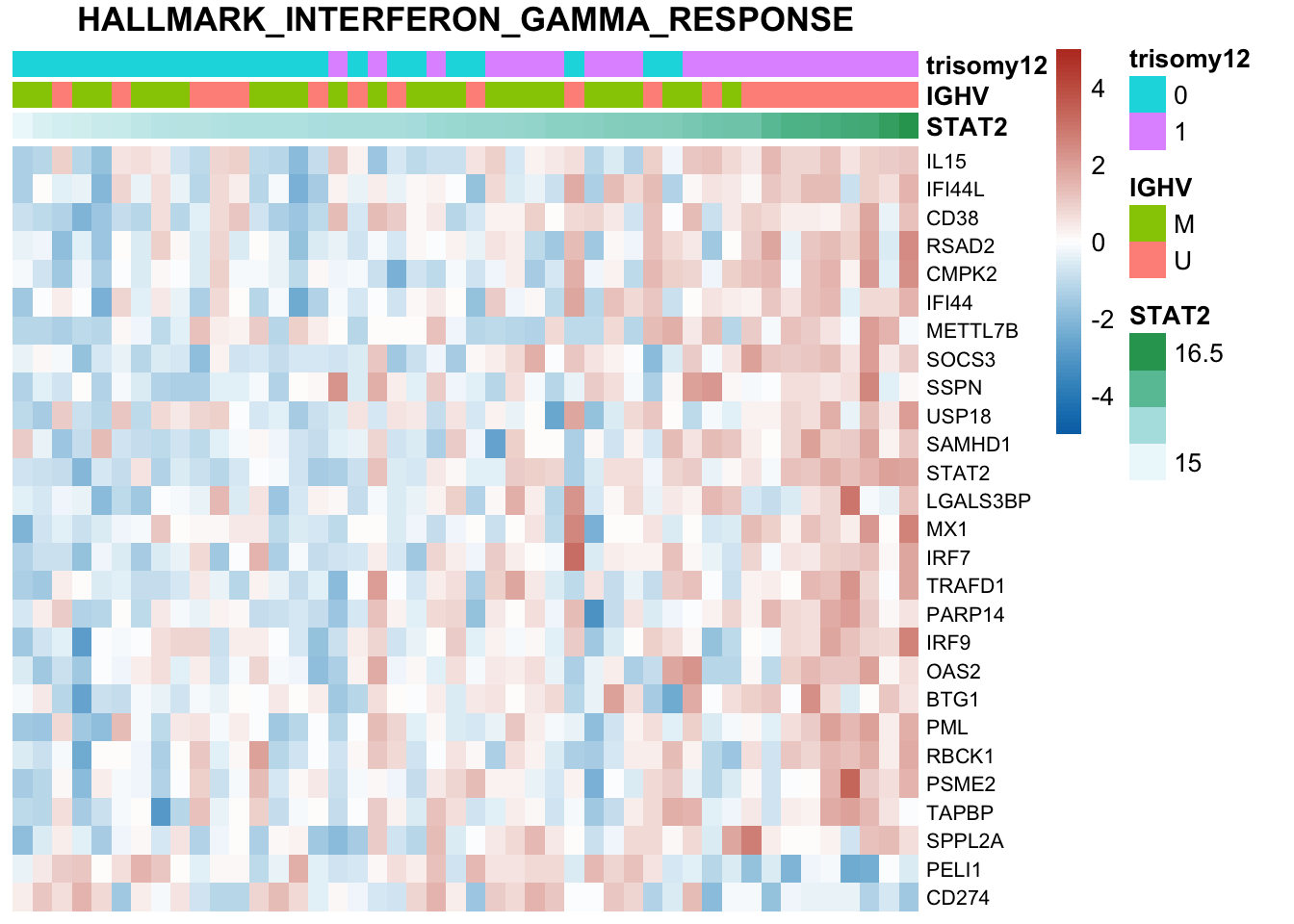
no blocking for IGHV or trisomy12
blocking for IGHV and trisomy12
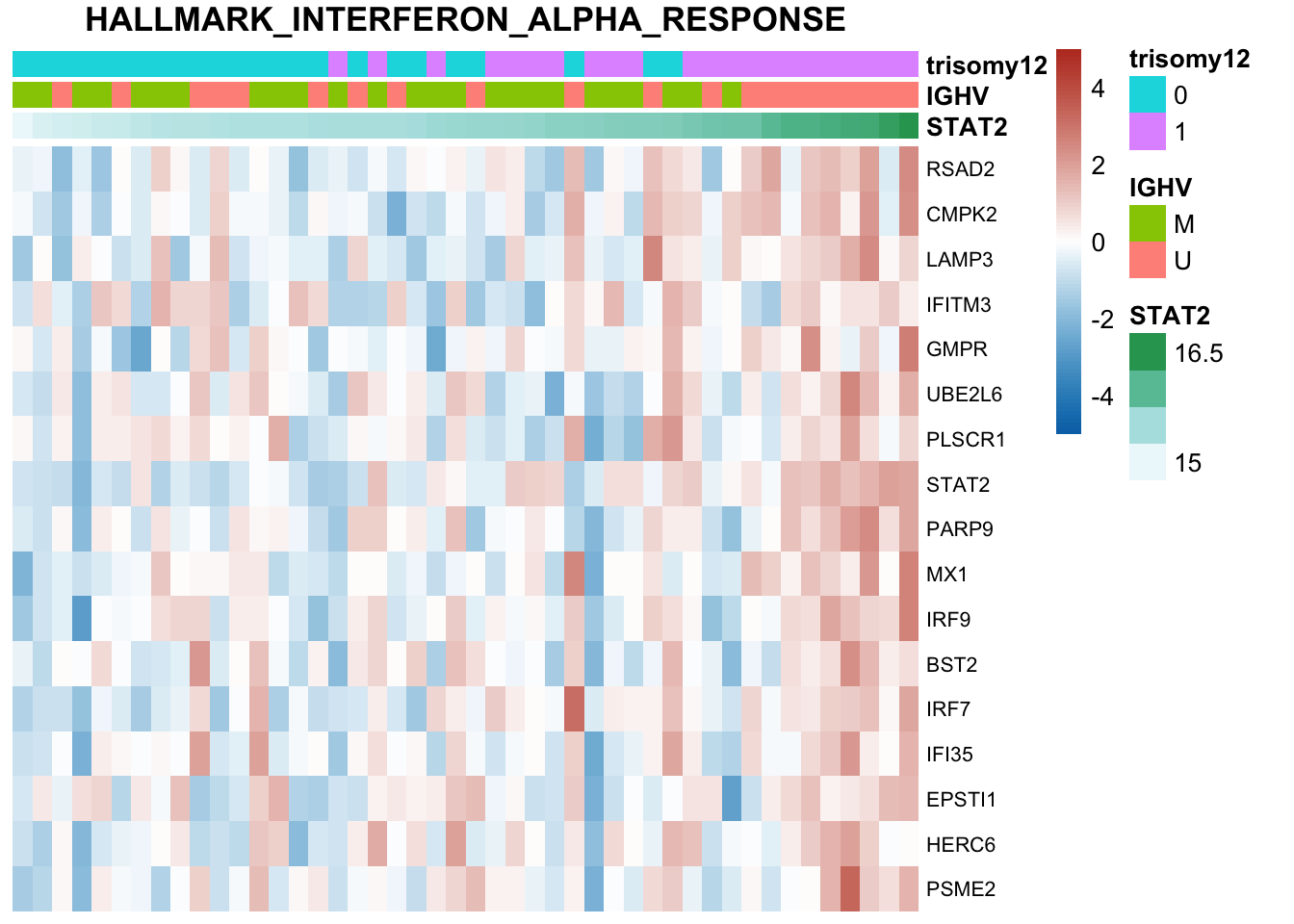
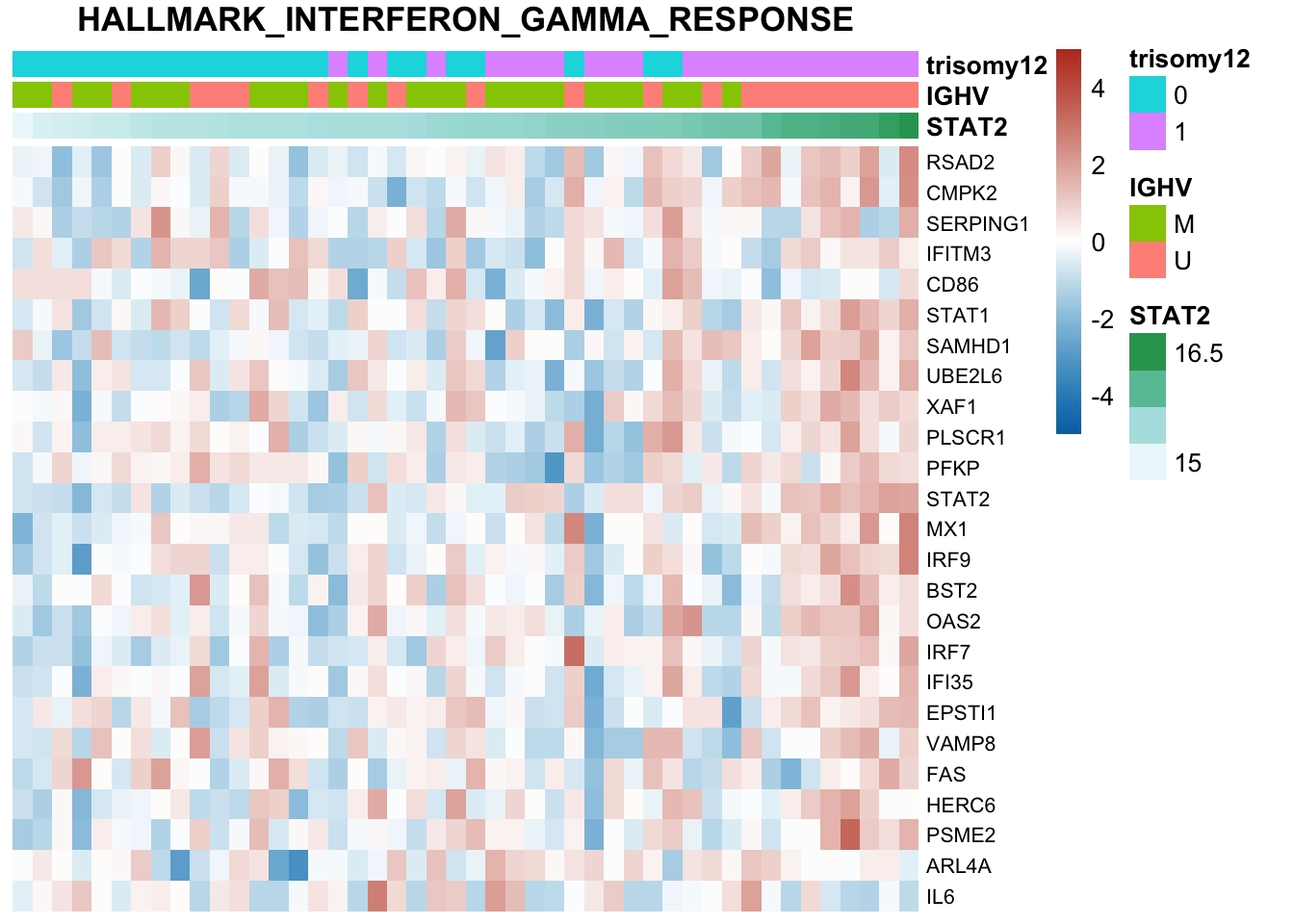
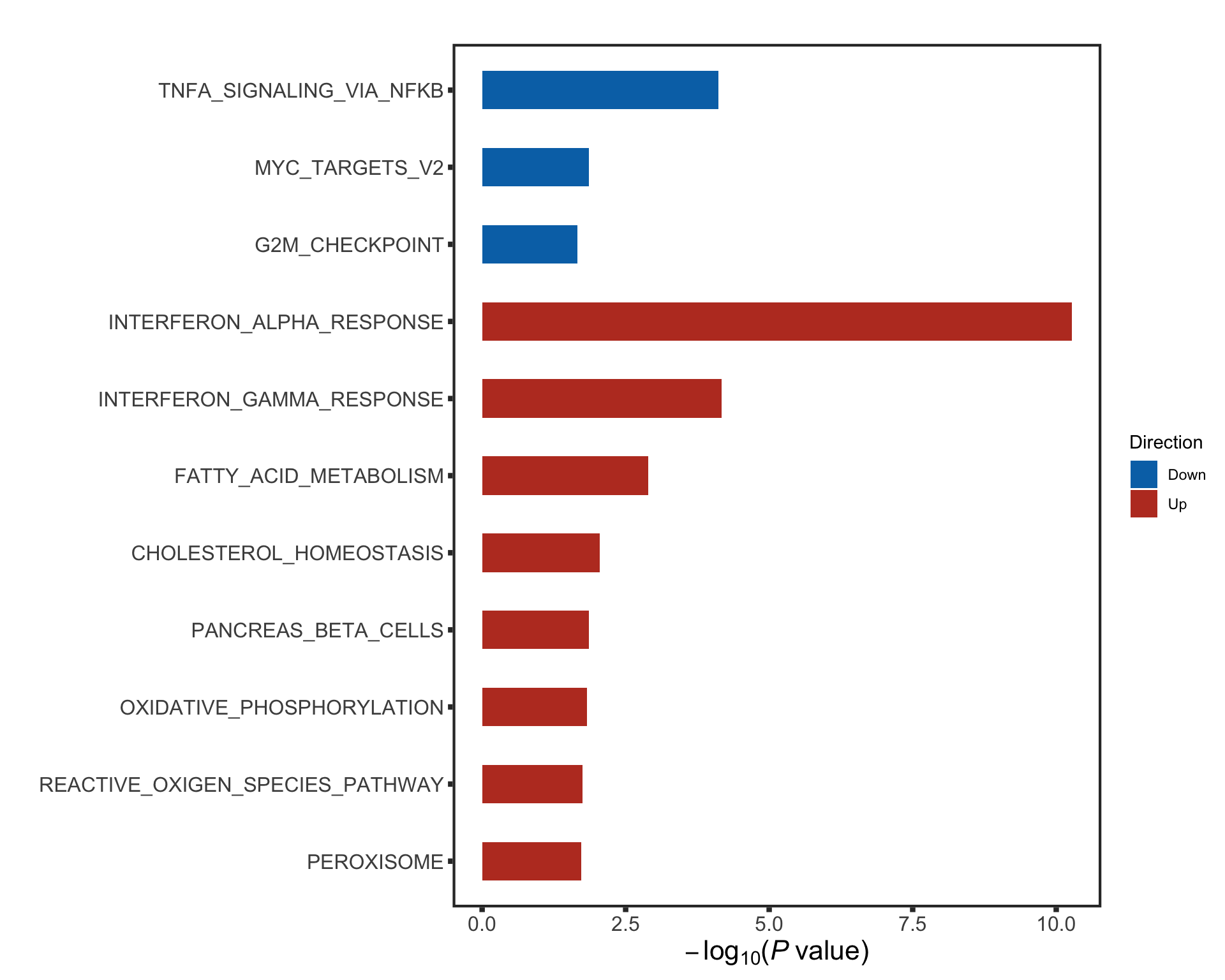 The pathways are similar as for no blocking
The pathways are similar as for no blocking
Pathway enrichment for protein expressions correlated with STAT2 protein level
Test
no blocking
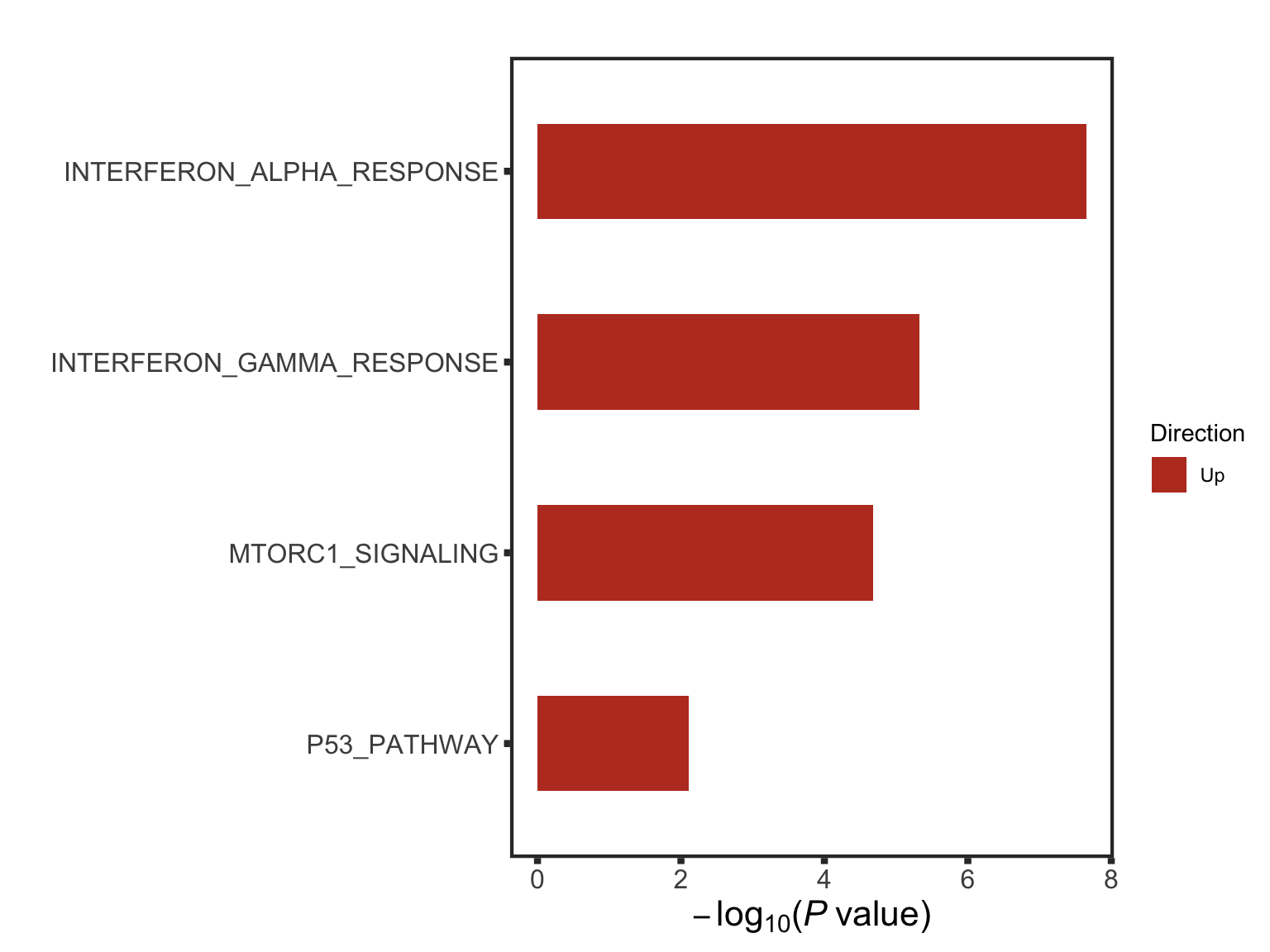
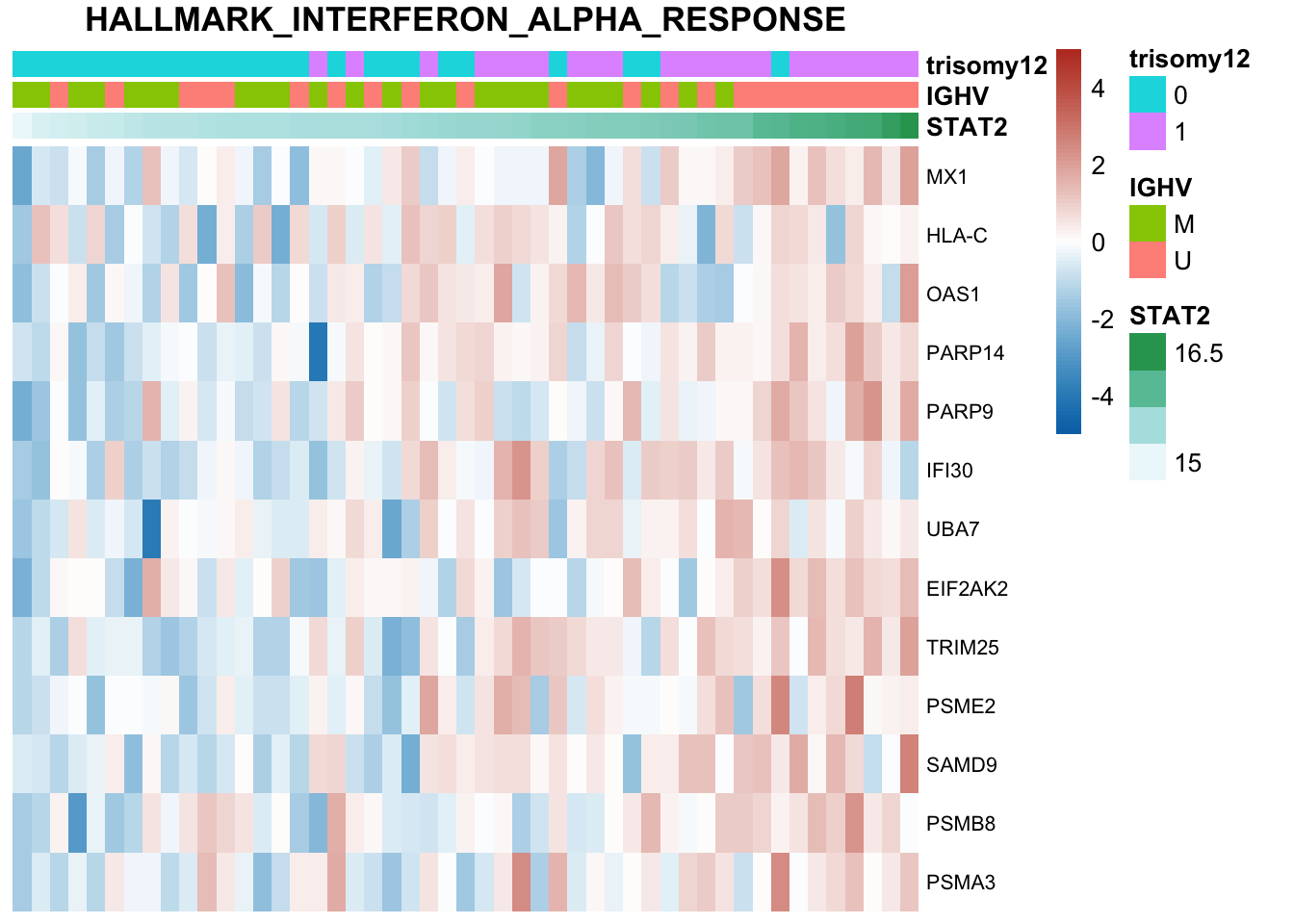
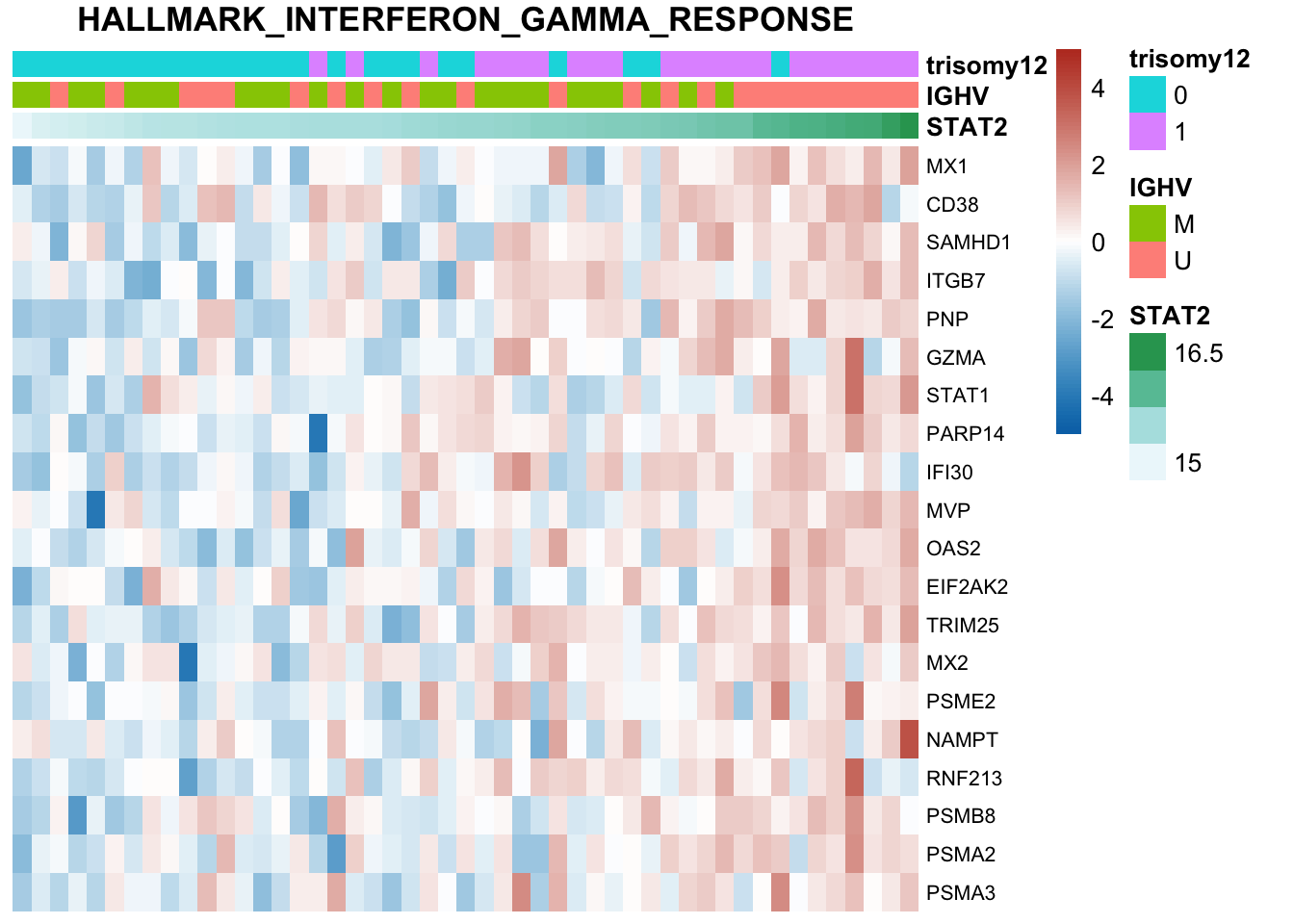
blocking for IGHV and trisomy12
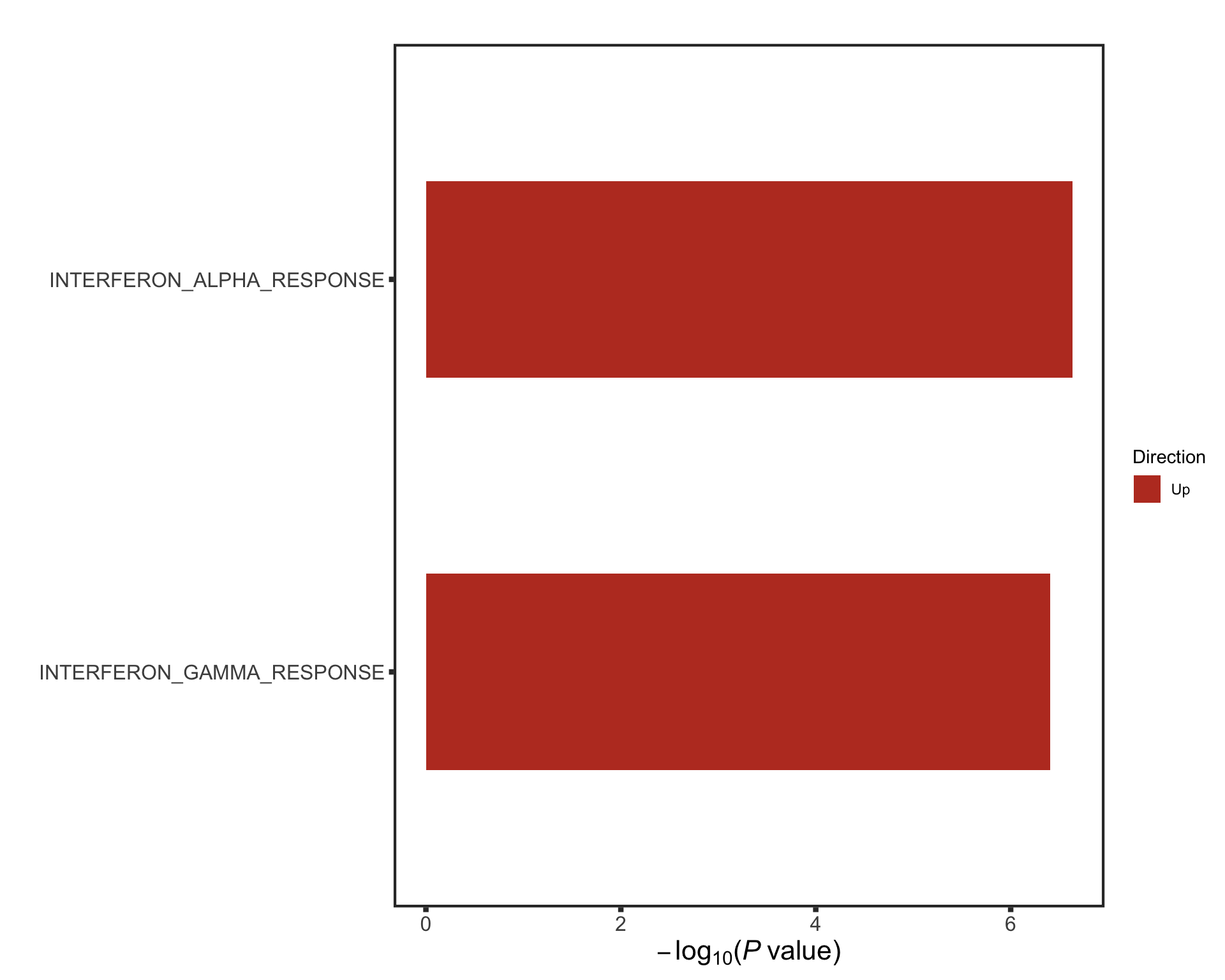
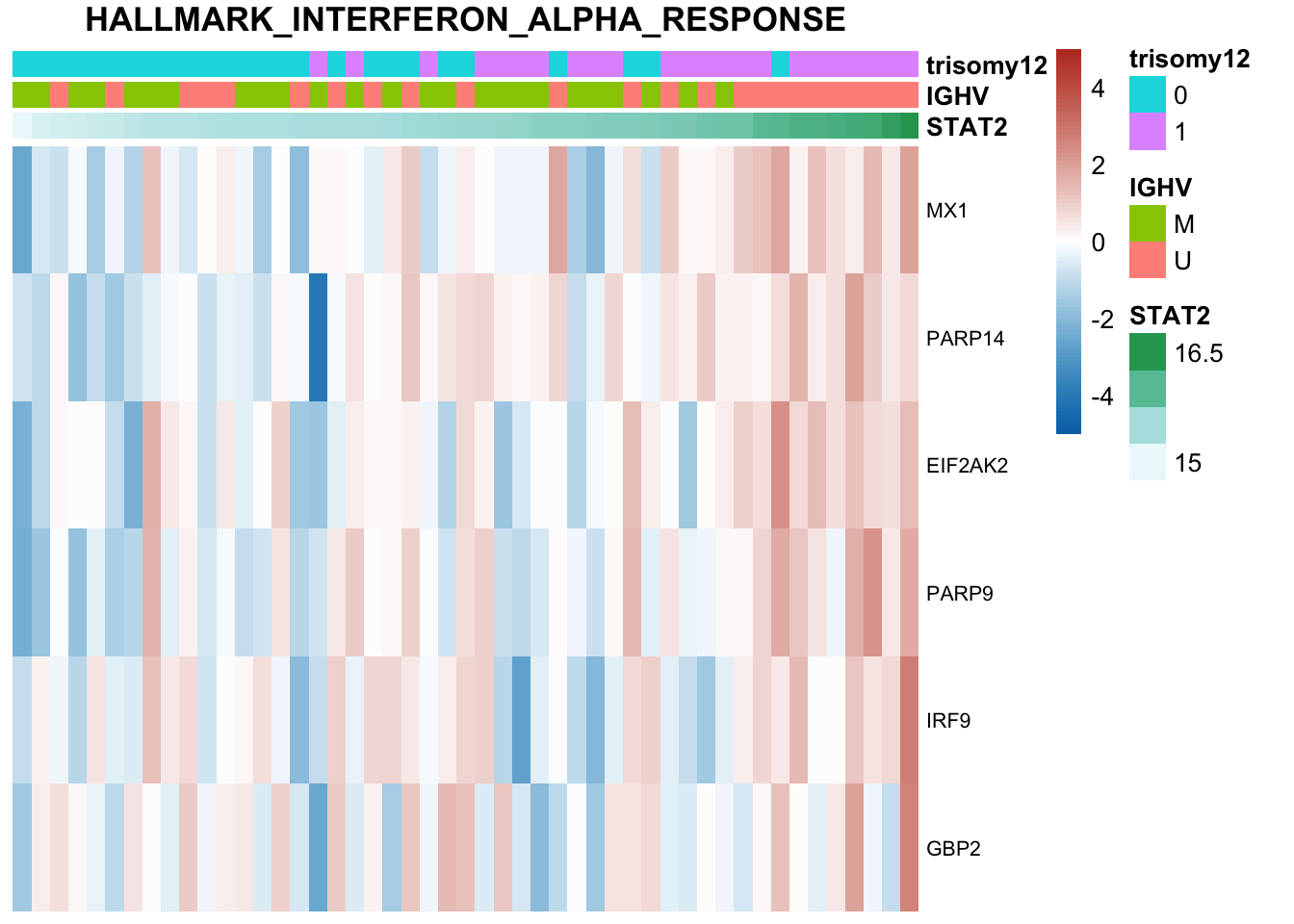
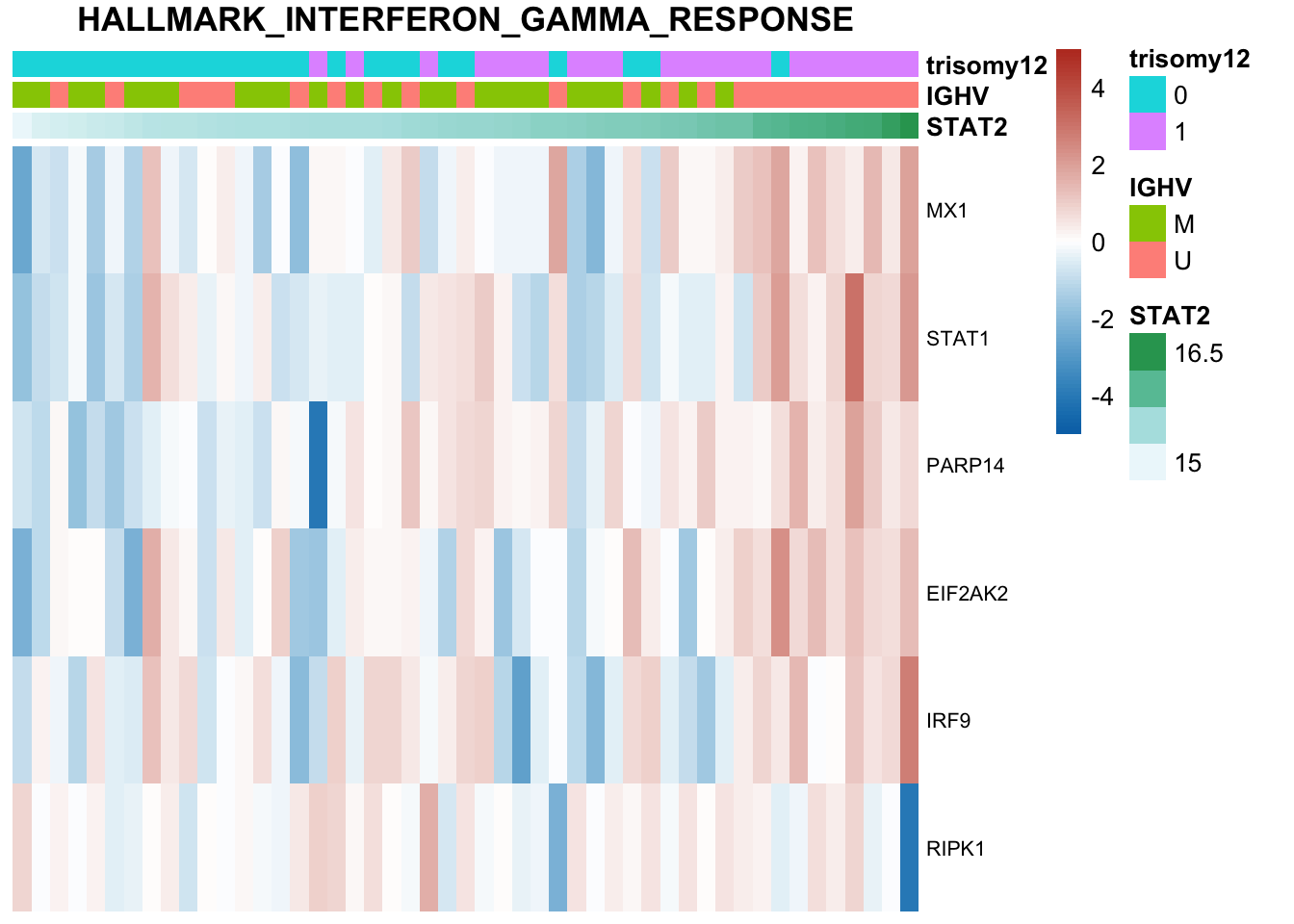
 Not many significant results when blocking for IGHV, suggesting most associations could be coufounded by IGHV and trisomy12
Not many significant results when blocking for IGHV, suggesting most associations could be coufounded by IGHV and trisomy12
R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DESeq2_1.26.0 latex2exp_0.4.0
[3] forcats_0.5.0 stringr_1.4.0
[5] dplyr_1.0.0 purrr_0.3.4
[7] readr_1.3.1 tidyr_1.1.0
[9] tibble_3.0.3 ggplot2_3.3.2
[11] tidyverse_1.3.0 SummarizedExperiment_1.16.1
[13] DelayedArray_0.12.3 BiocParallel_1.20.1
[15] matrixStats_0.56.0 Biobase_2.46.0
[17] GenomicRanges_1.38.0 GenomeInfoDb_1.22.1
[19] IRanges_2.20.2 S4Vectors_0.24.4
[21] BiocGenerics_0.32.0 glmnet_4.0-2
[23] Matrix_1.2-18 gtable_0.3.0
[25] limma_3.42.2 jyluMisc_0.1.5
[27] pheatmap_1.0.12 piano_2.2.0
[29] cowplot_1.0.0
loaded via a namespace (and not attached):
[1] shinydashboard_0.7.1 tidyselect_1.1.0 RSQLite_2.2.0
[4] AnnotationDbi_1.48.0 htmlwidgets_1.5.1 grid_3.6.0
[7] maxstat_0.7-25 munsell_0.5.0 codetools_0.2-16
[10] DT_0.14 withr_2.2.0 colorspace_1.4-1
[13] knitr_1.29 rstudioapi_0.11 ggsignif_0.6.0
[16] labeling_0.3 git2r_0.27.1 slam_0.1-47
[19] GenomeInfoDbData_1.2.2 KMsurv_0.1-5 farver_2.0.3
[22] bit64_0.9-7 rprojroot_1.3-2 vctrs_0.3.1
[25] generics_0.0.2 TH.data_1.0-10 xfun_0.15
[28] sets_1.0-18 R6_2.4.1 locfit_1.5-9.4
[31] bitops_1.0-6 fgsea_1.12.0 assertthat_0.2.1
[34] promises_1.1.1 scales_1.1.1 multcomp_1.4-13
[37] nnet_7.3-14 sandwich_2.5-1 workflowr_1.6.2
[40] rlang_0.4.7 genefilter_1.68.0 splines_3.6.0
[43] rstatix_0.6.0 acepack_1.4.1 broom_0.7.0
[46] checkmate_2.0.0 yaml_2.2.1 abind_1.4-5
[49] modelr_0.1.8 backports_1.1.8 httpuv_1.5.4
[52] Hmisc_4.4-0 tools_3.6.0 relations_0.6-9
[55] ellipsis_0.3.1 gplots_3.0.4 RColorBrewer_1.1-2
[58] Rcpp_1.0.5 base64enc_0.1-3 visNetwork_2.0.9
[61] zlibbioc_1.32.0 RCurl_1.98-1.2 ggpubr_0.4.0
[64] rpart_4.1-15 zoo_1.8-8 haven_2.3.1
[67] cluster_2.1.0 exactRankTests_0.8-31 fs_1.4.2
[70] magrittr_1.5 data.table_1.12.8 openxlsx_4.1.5
[73] reprex_0.3.0 survminer_0.4.7 mvtnorm_1.1-1
[76] hms_0.5.3 shinyjs_1.1 mime_0.9
[79] evaluate_0.14 xtable_1.8-4 XML_3.98-1.20
[82] rio_0.5.16 jpeg_0.1-8.1 readxl_1.3.1
[85] gridExtra_2.3 shape_1.4.4 compiler_3.6.0
[88] KernSmooth_2.23-17 crayon_1.3.4 htmltools_0.5.0
[91] later_1.1.0.1 Formula_1.2-3 geneplotter_1.64.0
[94] lubridate_1.7.9 DBI_1.1.0 dbplyr_1.4.4
[97] MASS_7.3-51.6 car_3.0-8 cli_2.0.2
[100] marray_1.64.0 gdata_2.18.0 igraph_1.2.5
[103] pkgconfig_2.0.3 km.ci_0.5-2 foreign_0.8-71
[106] xml2_1.3.2 foreach_1.5.0 annotate_1.64.0
[109] XVector_0.26.0 drc_3.0-1 rvest_0.3.5
[112] digest_0.6.25 rmarkdown_2.3 cellranger_1.1.0
[115] fastmatch_1.1-0 survMisc_0.5.5 htmlTable_2.0.1
[118] curl_4.3 shiny_1.5.0 gtools_3.8.2
[121] lifecycle_0.2.0 jsonlite_1.7.0 carData_3.0-4
[124] fansi_0.4.1 pillar_1.4.6 lattice_0.20-41
[127] fastmap_1.0.1 httr_1.4.1 plotrix_3.7-8
[130] survival_3.2-3 glue_1.4.1 zip_2.0.4
[133] png_0.1-7 iterators_1.0.12 bit_1.1-15.2
[136] stringi_1.4.6 blob_1.2.1 latticeExtra_0.6-29
[139] caTools_1.18.0 memoise_1.1.0