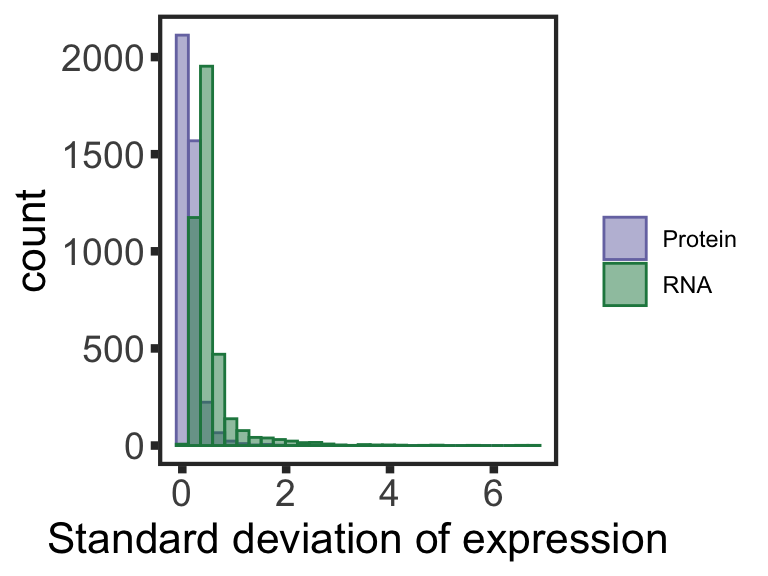
Analyzing buffering effect
Detect buffered and non-buffered proteins
Preprocessing protein and RNA data
#subset samples and genes
overSampe <- intersect(colnames(ddsCLL), colnames(protCLL))
overGene <- intersect(rownames(ddsCLL), rowData(protCLL)$ensembl_gene_id)
ddsSub <- ddsCLL[overGene, overSampe]
protSub <- protCLL[match(overGene, rowData(protCLL)$ensembl_gene_id),overSampe]
rowData(ddsSub)$uniprotID <- rownames(protSub)[match(rownames(ddsSub),rowData(protSub)$ensembl_gene_id)]
#vst
ddsSub.vst <- varianceStabilizingTransformation(ddsSub)
Differential expression on RNA level
ddsSub$trisomy12 <- patMeta[match(ddsSub$PatID,patMeta$Patient.ID),]$trisomy12
ddsSub$IGHV <- patMeta[match(ddsSub$PatID, patMeta$Patient.ID),]$IGHV.status
design(ddsSub) <- ~ trisomy12 + IGHV
deRes <- DESeq(ddsSub, betaPrior = TRUE)
rnaRes <- results(deRes, name = "trisomy121", tidy = TRUE) %>%
dplyr::rename(geneID = row, log2FC.rna = log2FoldChange,
pvalue.rna = pvalue, padj.rna = padj, stat.rna= stat) %>%
select(geneID, log2FC.rna, pvalue.rna, padj.rna, stat.rna)
Protein abundance changes related to trisomy12
fdrCut <- 0.1
protRes <- resList %>% filter(Gene == "trisomy12") %>%
dplyr::rename(uniprotID = id,
pvalue = P.Value, padj = adj.P.IHW,
chrom = Chr) %>%
mutate(geneID = rowData(protCLL[uniprotID,])$ensembl_gene_id) %>%
select(name, uniprotID, geneID, chrom, logFC, pvalue, padj, t) %>%
dplyr::rename(stat =t) %>%
arrange(pvalue) %>% as_tibble()
Combine
allRes <- left_join(protRes, rnaRes, by = "geneID")
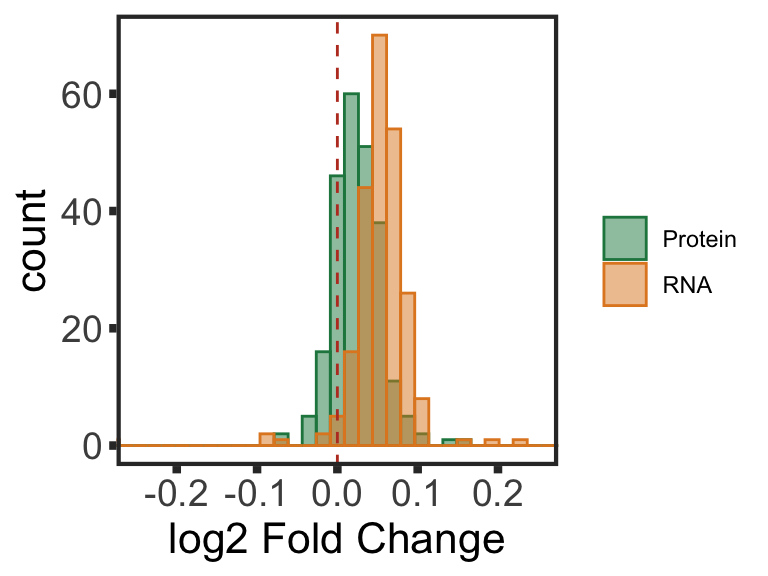
Only chr12 genes that are up-regulated are considered. Otherwise it's hard to intepret the dosage effect.
bufferTab <- allRes %>% filter(chrom %in% 12,stat.rna > 0) %>%
ungroup() %>%
mutate(stat.prot.sqrt = sqrt(stat),
stat.prot.center = stat.prot.sqrt - mean(stat.prot.sqrt)) %>%
mutate(score = -stat.prot.center*stat.rna) %>%
mutate(ifBuffer = case_when(
padj < 0.1 & padj.rna < 0.1 & stat > 0 ~ "non-Buffered",
padj > 0.1 & padj.rna < 0.1 ~ "Buffered",
padj < 0.1 & padj.rna > 0.1 & stat > 0 ~ "Enhanced",
TRUE ~ "Undetermined"
)) %>%
arrange(desc(score))
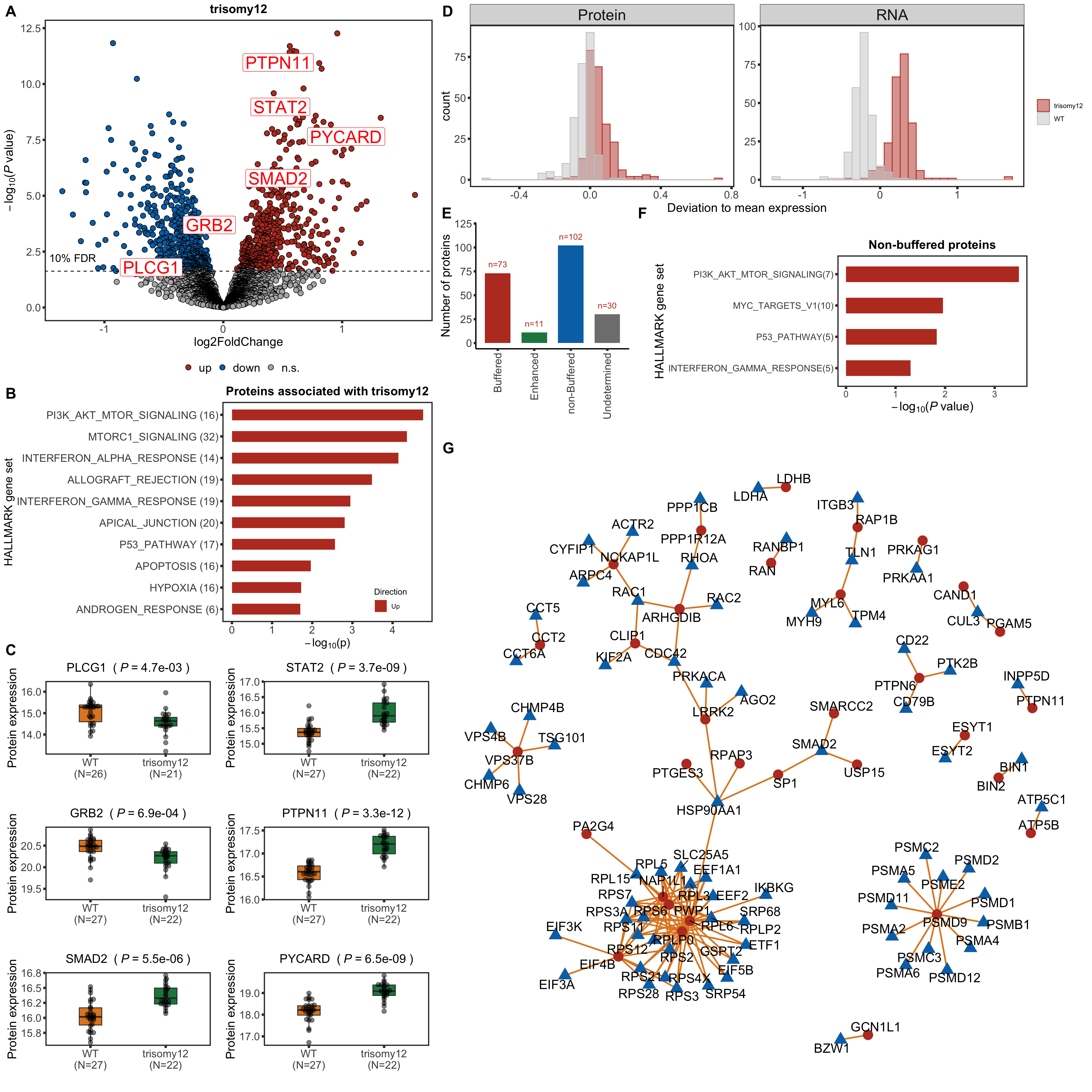
Summary plot
sumTab <- bufferTab %>% group_by(ifBuffer) %>%
summarise(n = length(name))
bufferPlot <- ggplot(sumTab, aes(x=ifBuffer, y = n)) +
geom_bar(aes(fill = ifBuffer), stat="identity", width = 0.7) +
geom_text(aes(label = paste0("n=", n)),vjust=-1,col=colList[1]) +
scale_fill_manual(values =c(Buffered = colList[1],
Enhanced = colList[4],
`non-Buffered` = colList[2],
Undetermined = "grey50")) +
theme_half + theme(axis.text.x = element_text(angle = 90, hjust=1, vjust=0.5),
legend.position = "none") +
ylab("Number of proteins") + ylim(0,120) +xlab("")
bufferPlot
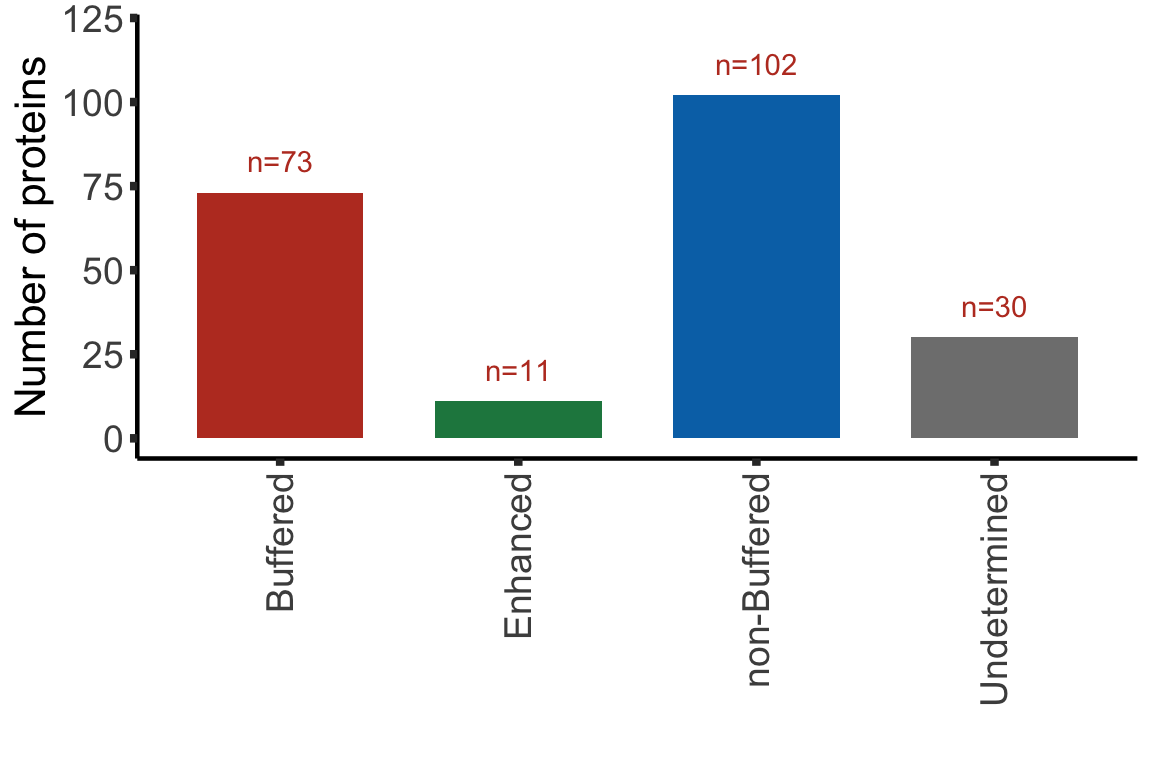
Plot example cases of buffered and non-buffered proteins
protList <- c("PTPN11","DDX51","BCAT1", "METTL7A")
geneList <- bufferTab[match(protList, bufferTab$name),]$geneID
pList <- lapply(geneList, function(i) {
tabProt <- allProtTab %>% filter(id == i) %>%
select(id, patID, symbol,expr) %>% dplyr::rename(protExpr = expr)
tabRna <- allRnaTab %>% filter(id == i) %>%
select(id, patID, expr) %>% dplyr::rename(rnaExpr = expr)
plotTab <- left_join(tabProt, tabRna, by = c("id","patID")) %>%
filter(!is.na(protExpr), !is.na(rnaExpr)) %>%
mutate(trisomy12 = patMeta[match(patID, patMeta$Patient.ID),]$trisomy12) %>%
mutate(trisomy12 = ifelse(trisomy12 %in% 1, "yes","no"))
p <- ggplot(plotTab, aes(x=rnaExpr, y = protExpr)) +
geom_point(aes(col=trisomy12)) +
geom_smooth(formula = y~x, method="lm",se=FALSE, color = "grey50", linetype ="dashed" ) +
ggtitle(unique(plotTab$symbol)) +
ylab("Protein expression") + xlab("RNA expression") +
scale_color_manual(values =c(yes = colList[1],no=colList[2])) +
theme_full + theme(legend.position = "bottom")
ggExtra::ggMarginal(p, type = "histogram", groupFill = TRUE)
})
cowplot::plot_grid(plotlist = pList, ncol=2)
 In the current analysis, I removed all the proteins that can not be unqiuely mapped. Unfortunately SLC2A14 is one of them. Can we use another example for the enhanced proteins? Like METTL7A shown here. It's a methyltransferase that has been shown to be related to innate immunity.
In the current analysis, I removed all the proteins that can not be unqiuely mapped. Unfortunately SLC2A14 is one of them. Can we use another example for the enhanced proteins? Like METTL7A shown here. It's a methyltransferase that has been shown to be related to innate immunity.
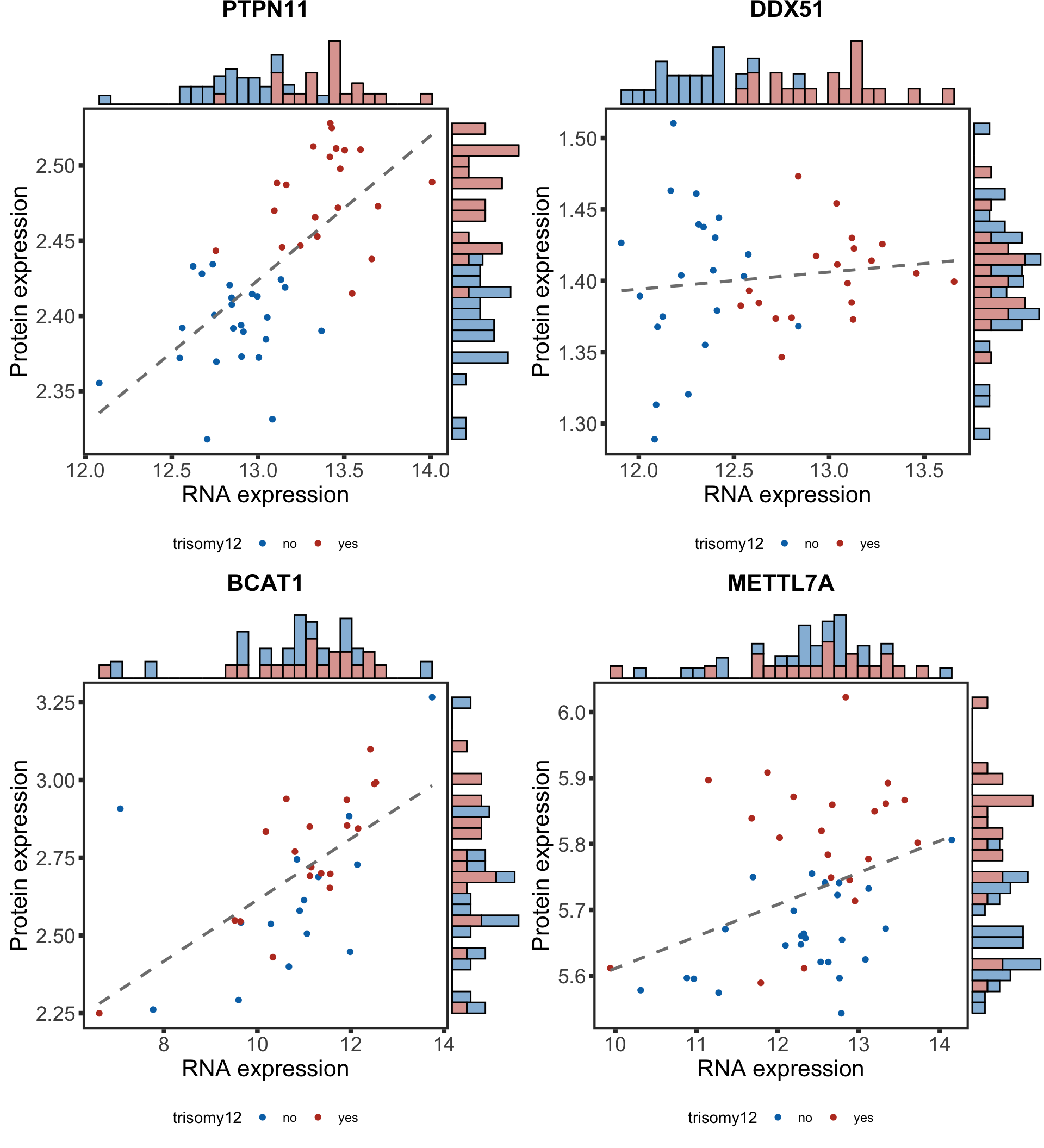
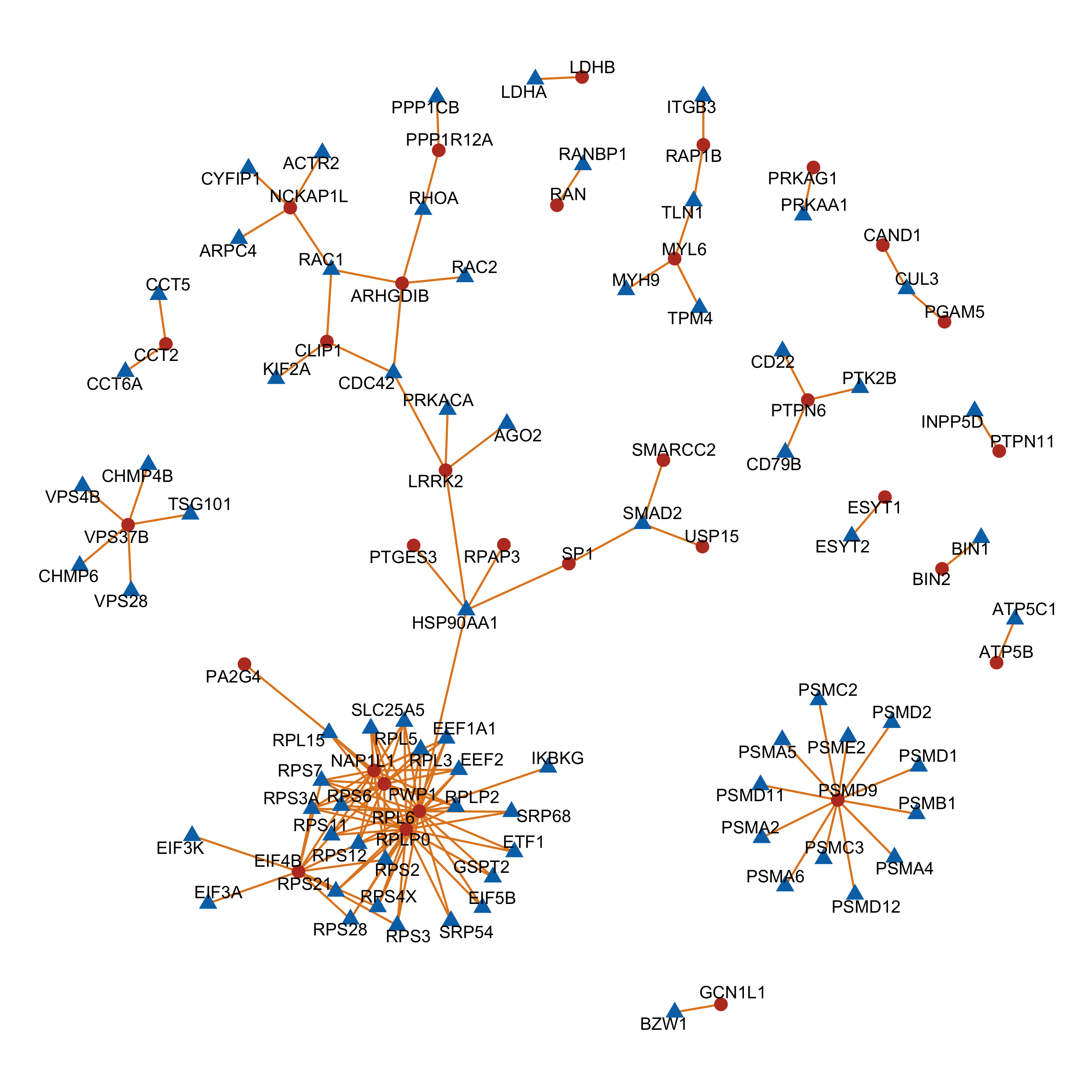
Enrichment of buffer and non-buffered proteins
Non-buffered prpteins
protList <- filter(bufferTab, ifBuffer == "non-Buffered")$name
refList <- unique(protExprTab$symbol)
enRes <- runFisher(protList, refList, gmts$H, pCut =0.05, ifFDR = FALSE,removePrefix = "HALLMARK_",
plotTitle = "Non-buffered proteins", insideLegend = TRUE,
setName = "HALLMARK gene set")
bufferEnrich <- enRes$enrichPlot + theme(plot.margin = margin(1,3,1,1, unit = "cm"))
bufferEnrich
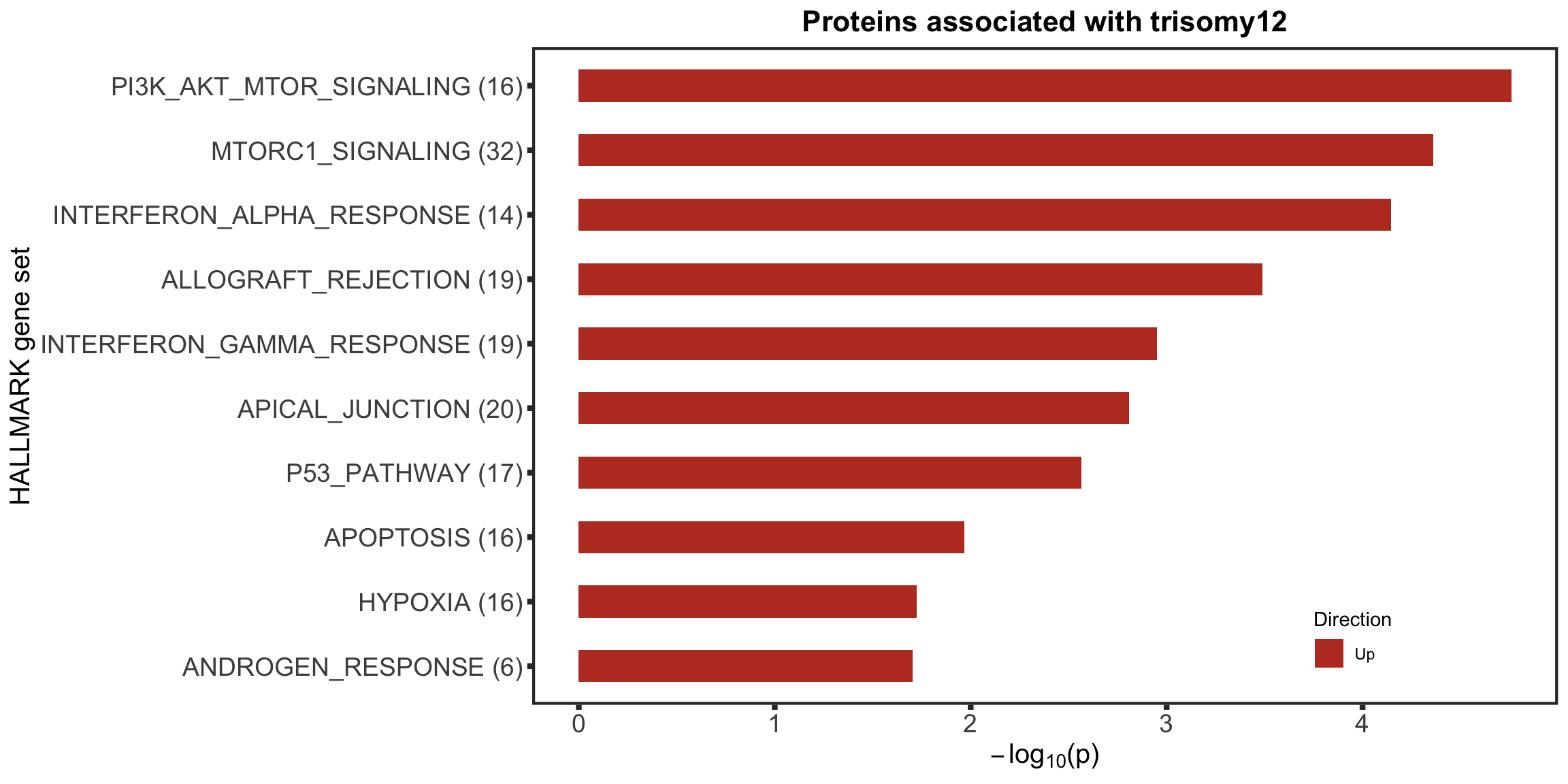
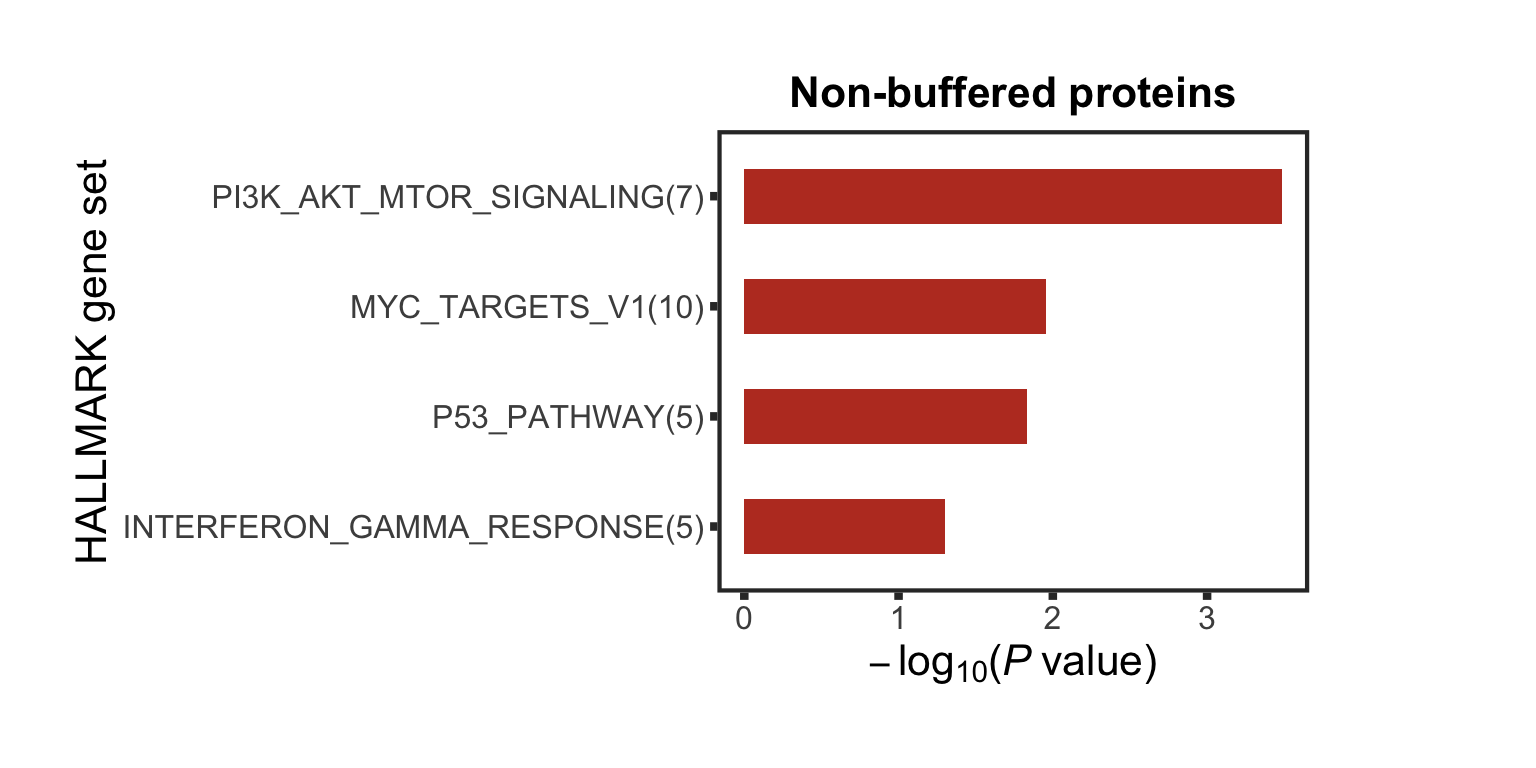 Those pathways passed p <0.05, but only PI3K_ATK_MTOR passed 10% FDR.
Those pathways passed p <0.05, but only PI3K_ATK_MTOR passed 10% FDR.
The result is a little different to the one ealier. Because a different enrichment method is used here, as we are not using the 'buffering score' in the manuscript.
Buffered proteins
protList <- filter(bufferTab, ifBuffer == "Buffered")$name
enRes <- runFisher(protList, refList, gmts$H, pCut =0.05, ifFDR = FALSE)
[1] "No sets passed the criteria"
No enrichment
Plot expression on genomic coordiate
load("../output/exprCNV_enc.RData")
patBack <- dplyr::filter(patMeta, Patient.ID %in% unique(allProtTab$patID)) %>%
dplyr::select(Patient.ID, trisomy12) %>%
dplyr::rename(patID = Patient.ID) %>%
mutate_all(as.character) %>%
mutate_at(vars(-patID),str_replace, "1","yes") %>%
mutate_at(vars(-patID),str_replace, "0","no")
plotExprVar <- function(gene, chr, patBack, allBand, allLine, allProtTab, allRnaTab, protLine = NULL,
region = c(-Inf,Inf),ifTrend = FALSE, normalize = TRUE, maxVal =2, minVal=-2) {
#table for cyto band
bandTab <- filter(allBand, ChromID == chr, chromStart >= region[1], chromEnd <= region[2]) %>%
mutate(chromMid = chromMid)
#table for expression
plotProtTab <- filter(allProtTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
plotRnaTab <- filter(allRnaTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
#summarise group mean
plotProtTab <- plotProtTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
plotRnaTab <- plotRnaTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
if (!is.null(protLine)) {
bufferLineTab <- plotProtTab %>%
select(symbol, mid_position, meanExpr, group) %>%
filter(symbol %in% protLine) %>%
pivot_wider(names_from = group, values_from = meanExpr) %>%
mutate(lowVal = map2_dbl(yes, no, min),
highVal = map2_dbl(yes, no, max))
} else bufferLineTab <- NULL
xMax <- max(bandTab$chromEnd, na.rm = T)
#main plot for Protein
gPro <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal+0.5, ymax=maxVal-0.5,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal-0.5, hjust =1, angle = 90, size=2.5)
if (!is.null(protLine)) {
gPro <- gPro + geom_segment(data = bufferLineTab, aes(x=mid_position, xend = mid_position,
y=lowVal, yend = highVal), linetype = "dashed")
}
gPro <- gPro + geom_rect(data = plotProtTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression (normalized by length)") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
yes = "darkred", no = "darkgreen")) +
scale_color_manual(values = c(yes = "darkred",no = "darkgreen")) +
ggtitle(paste0("Protein expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gPro <- gPro + geom_smooth(data =filter(plotProtTab, expr >0),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.5,
size =0.2, alpha=0.5)
}
#main plot for RNA
gRna <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal, ymax=maxVal,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal, hjust =1, angle = 90, size=2.5) +
geom_rect(data = plotRnaTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression Z-score") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
yes = "darkred", no = "darkgreen")) +
scale_color_manual(values = c(yes = "darkred", no = "darkgreen")) +
ggtitle(paste0("RNA expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gRna <- gRna + geom_smooth(data =filter(plotRnaTab),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.2,
size =0.2, alpha=0.5)
}
#for legend
## if the patient has CNV data
lgTab <- tibble(x= seq(6),y=seq(6),
Expression = c(rep("yes",3), rep("no",3)))
lg <- ggplot(lgTab, aes(x=x,y=y)) +
geom_point(aes(fill = Expression), shape =22,size=3) +
scale_fill_manual(values = c(yes = "darkred", no = "darkgreen"), name = gene) +
theme(legend.position = "bottom")
lg <- get_legend(lg)
return(list(plotPro = gPro, plotRNA = gRna, legend = lg))
}
Normalize protein and RNA expression
normalized <- TRUE
#if perform normalization
if (normalized) {
#for protein
exprMat <- select(allProtTab,patID, id,expr) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allProtTab <- select(allProtTab, -expr) %>% left_join(normTab, by = c("patID","id"))
#for RNA
exprMat <- select(allRnaTab,patID, id,expr) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allRnaTab <- select(allRnaTab, -expr) %>% left_join(normTab, by = c("patID","id"))
}
#pdf("./trisomy12_norm.pdf",height = 8, width = 10)
g <- plotExprVar("trisomy12","chr12",patBack,allBand, allLine,
allProtTab, allRnaTab, ifTrend = TRUE)
plot_grid(g$plotRNA, g$plotPro, g$legend, ncol = 1, rel_heights = c(1,1,0.2))
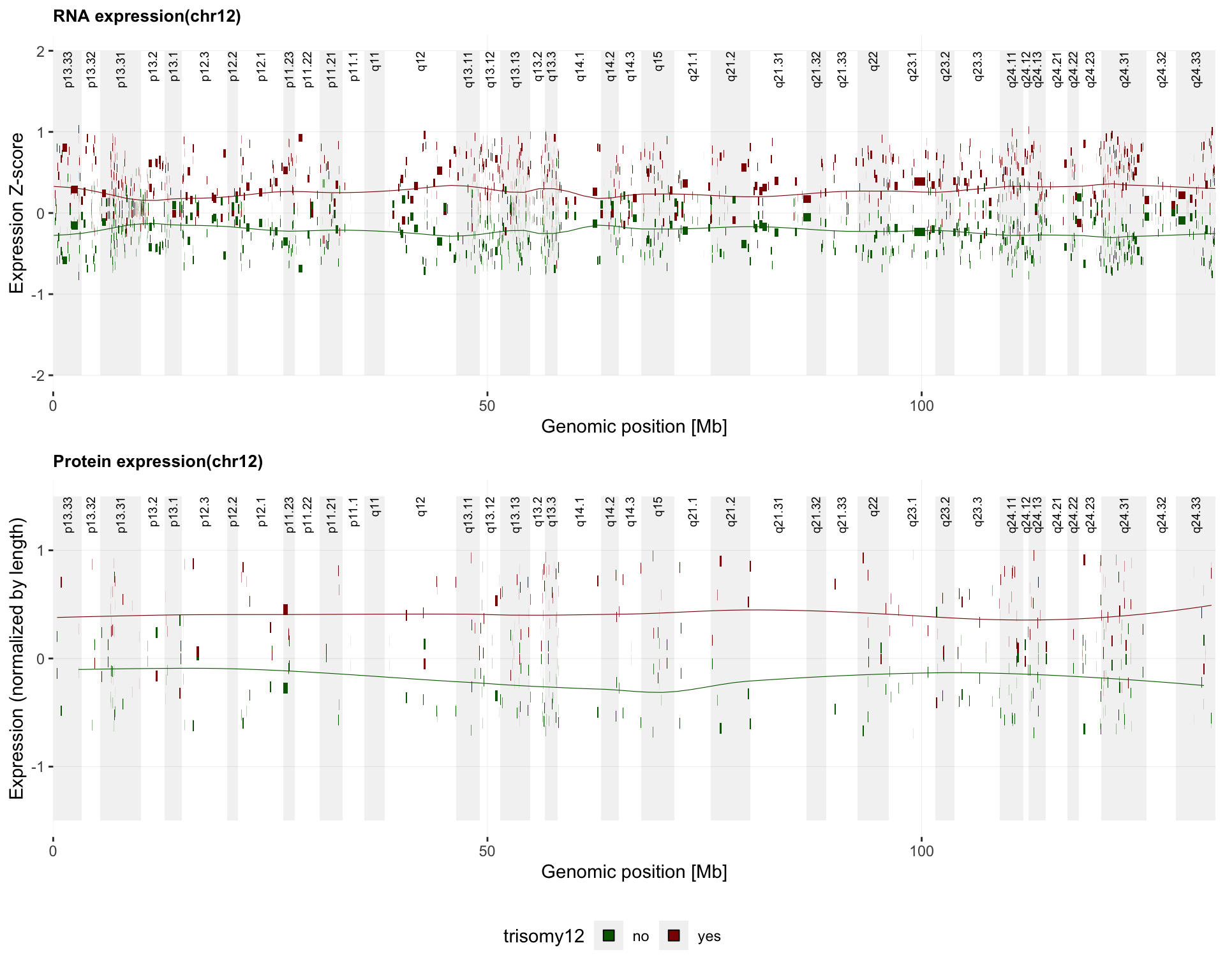
#dev.off()
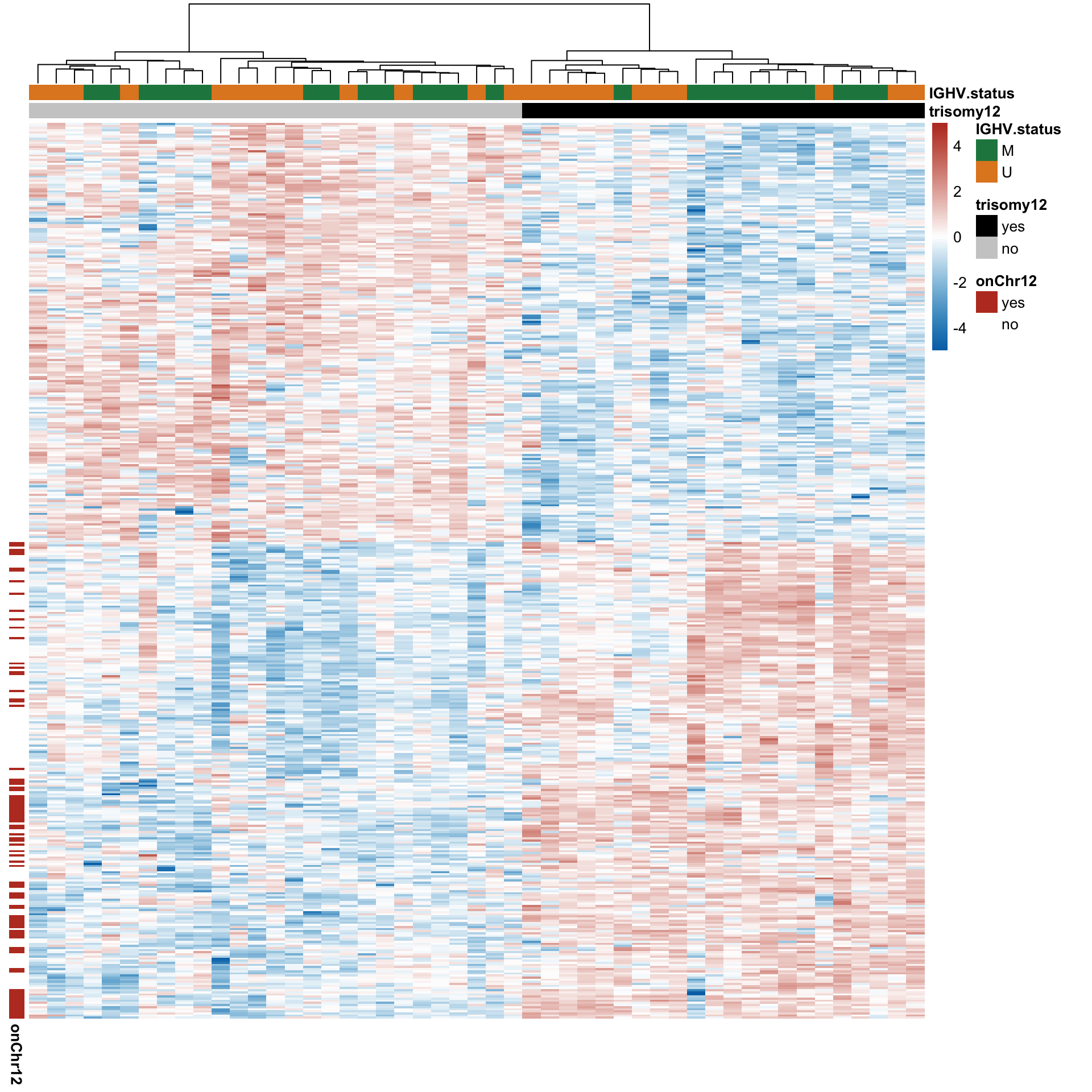
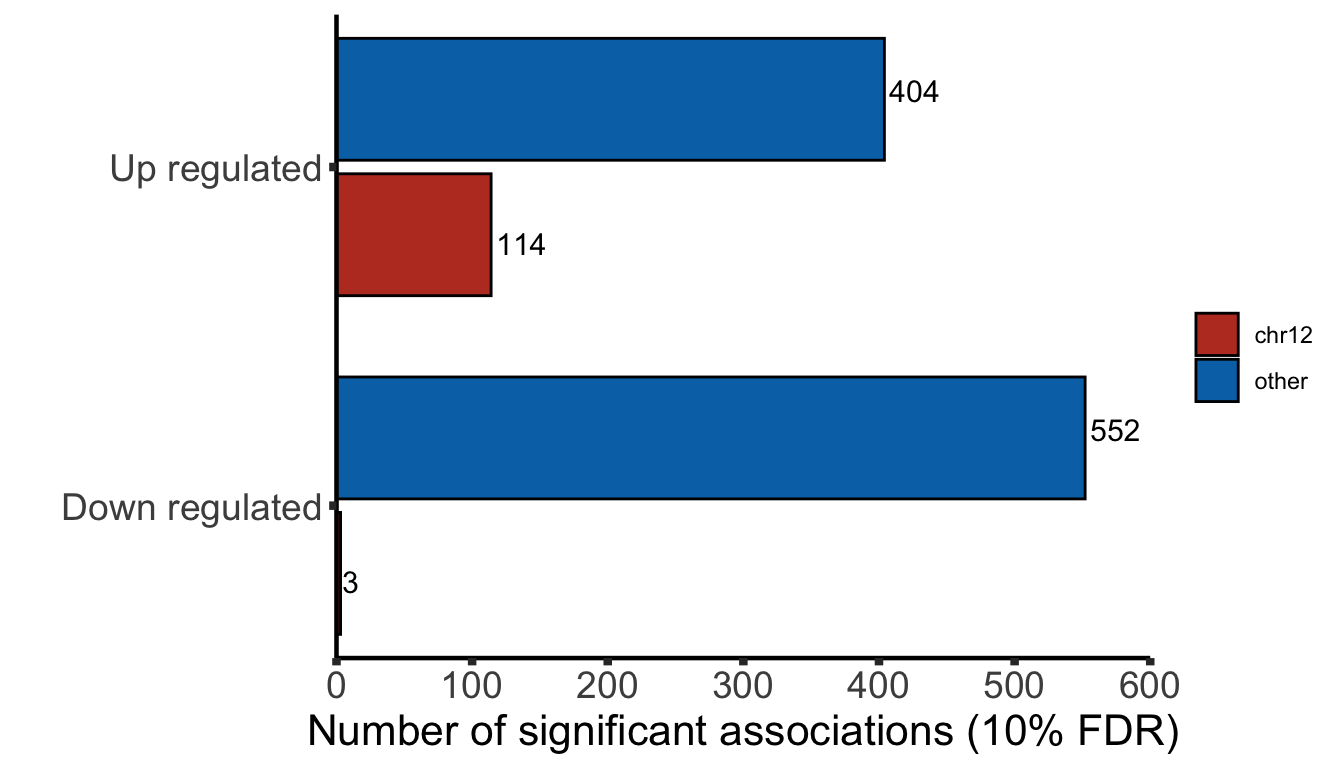
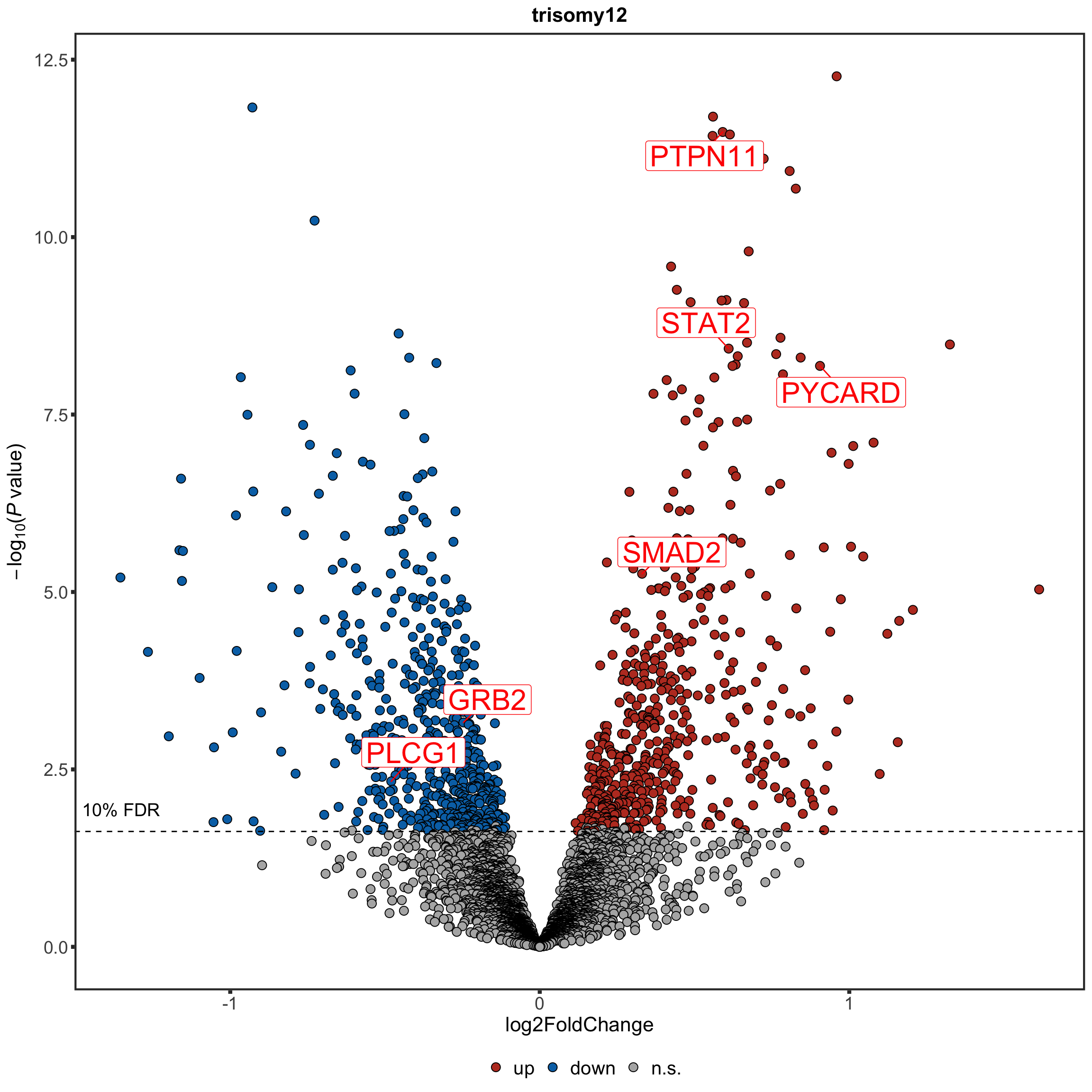
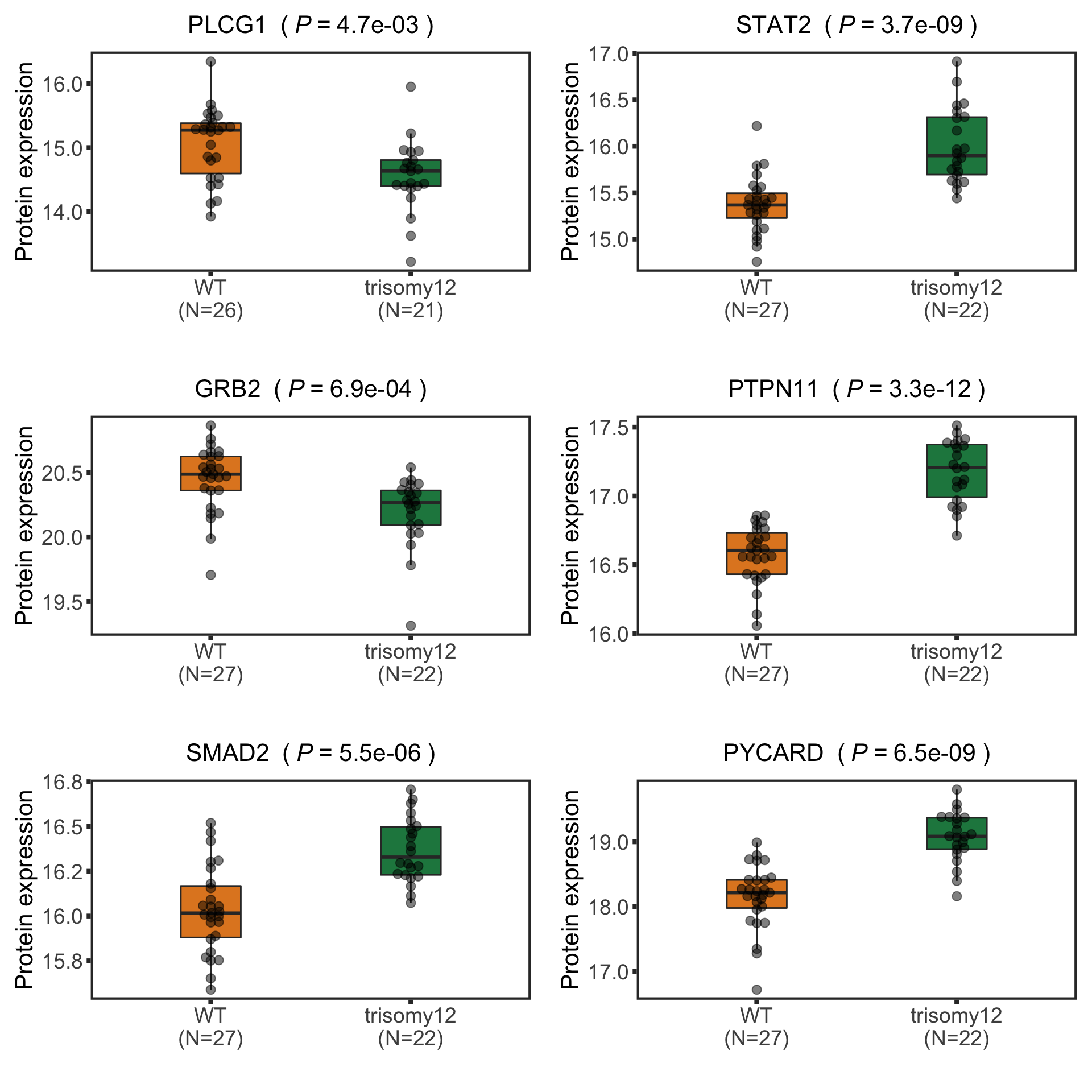
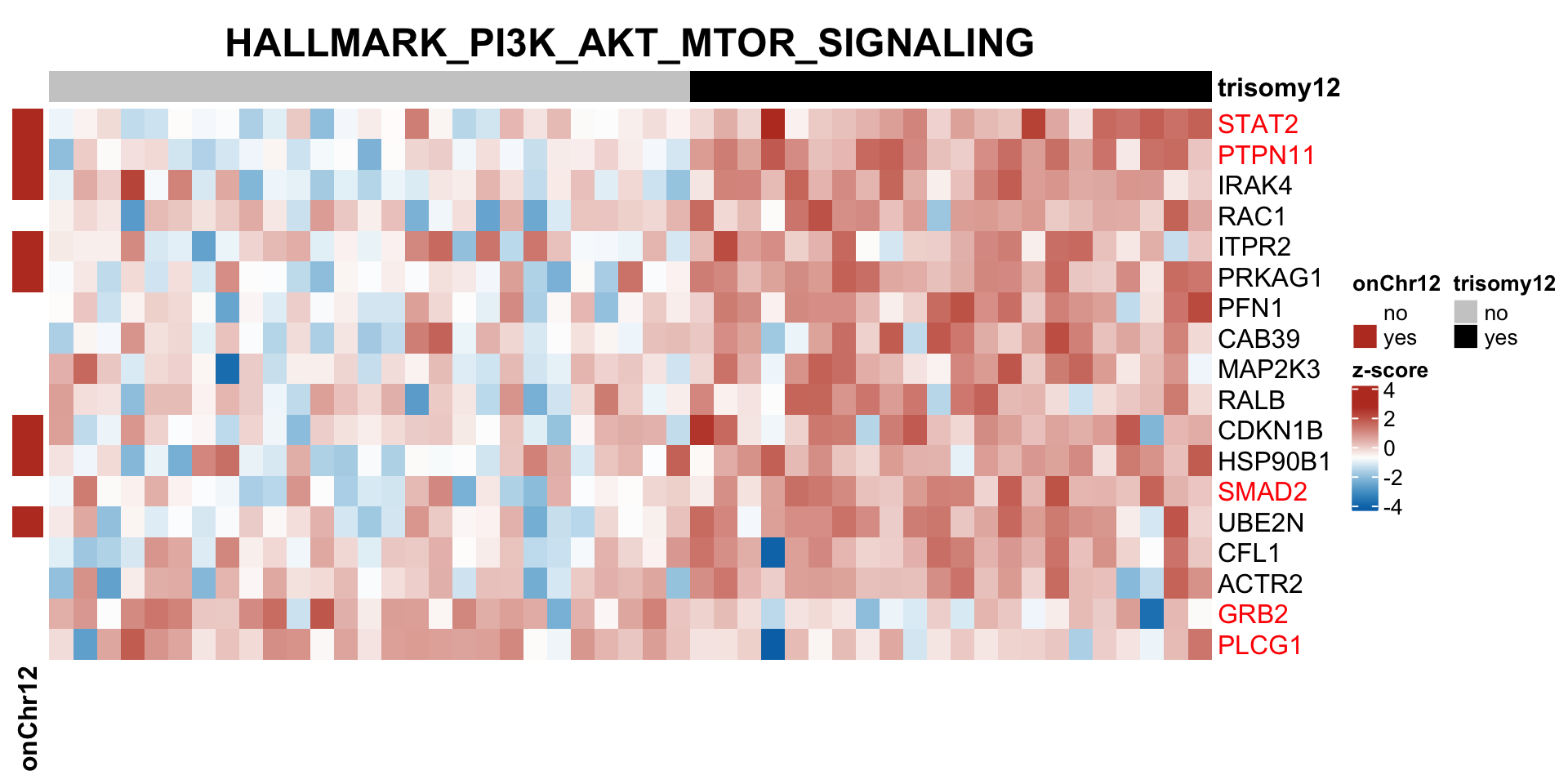
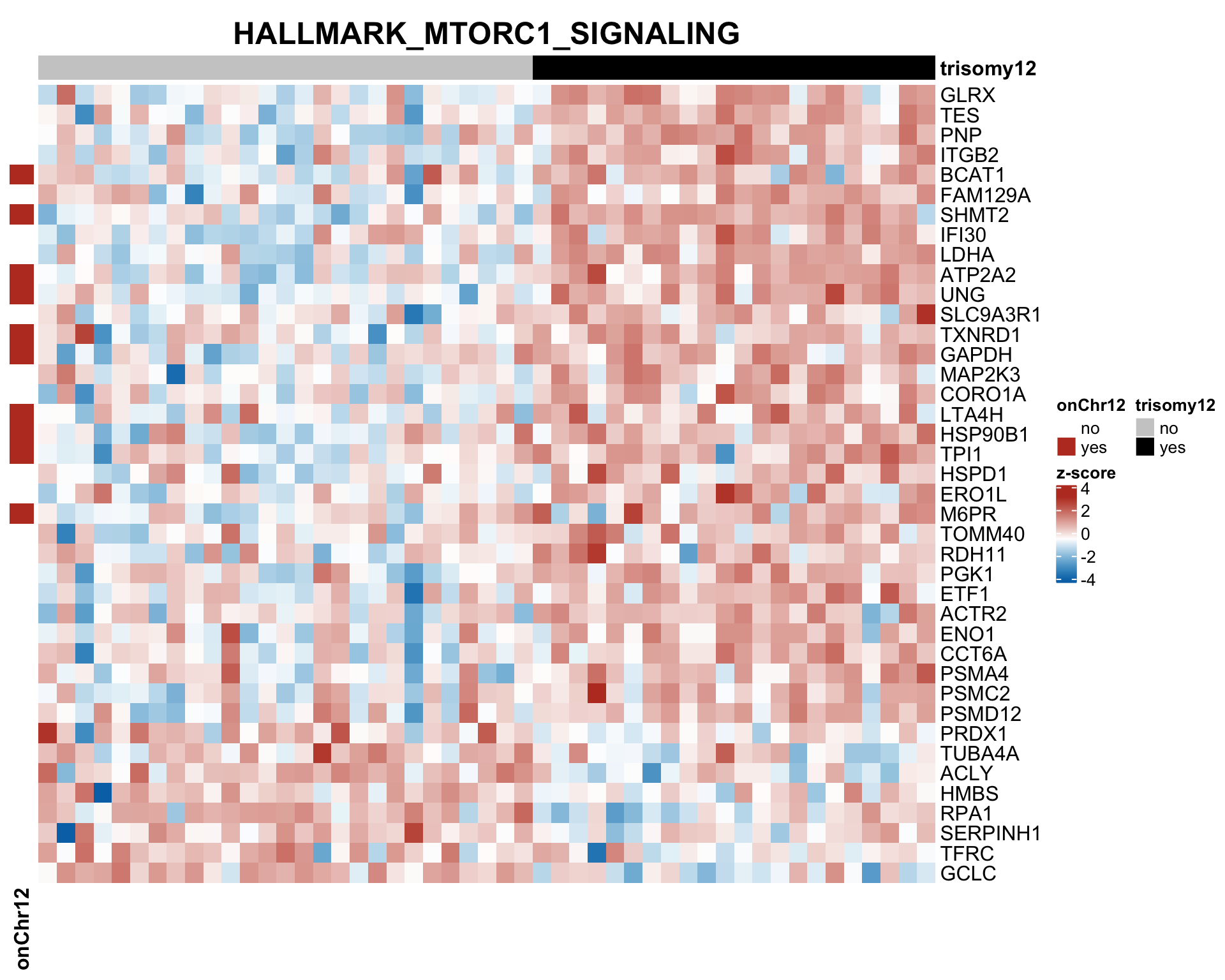
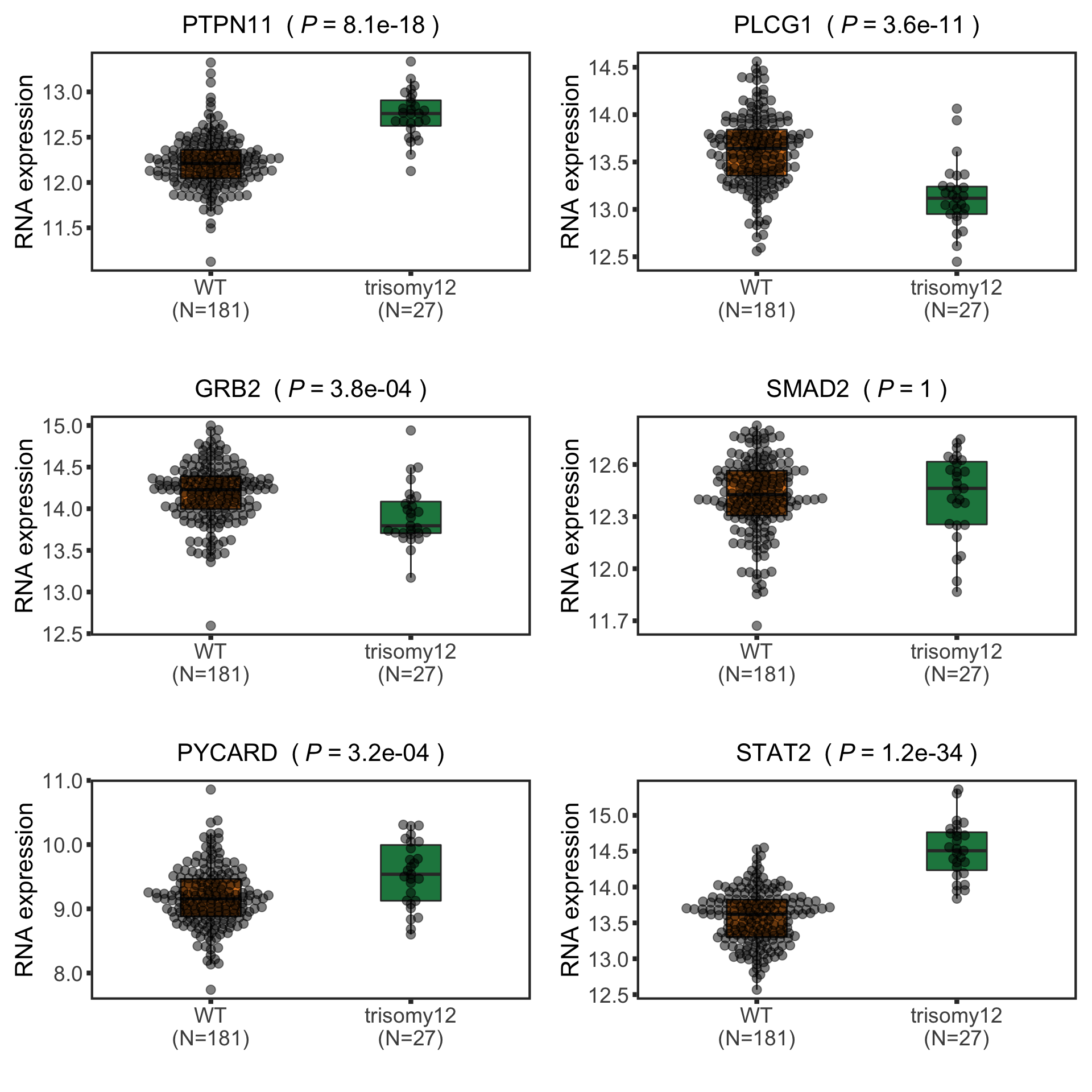
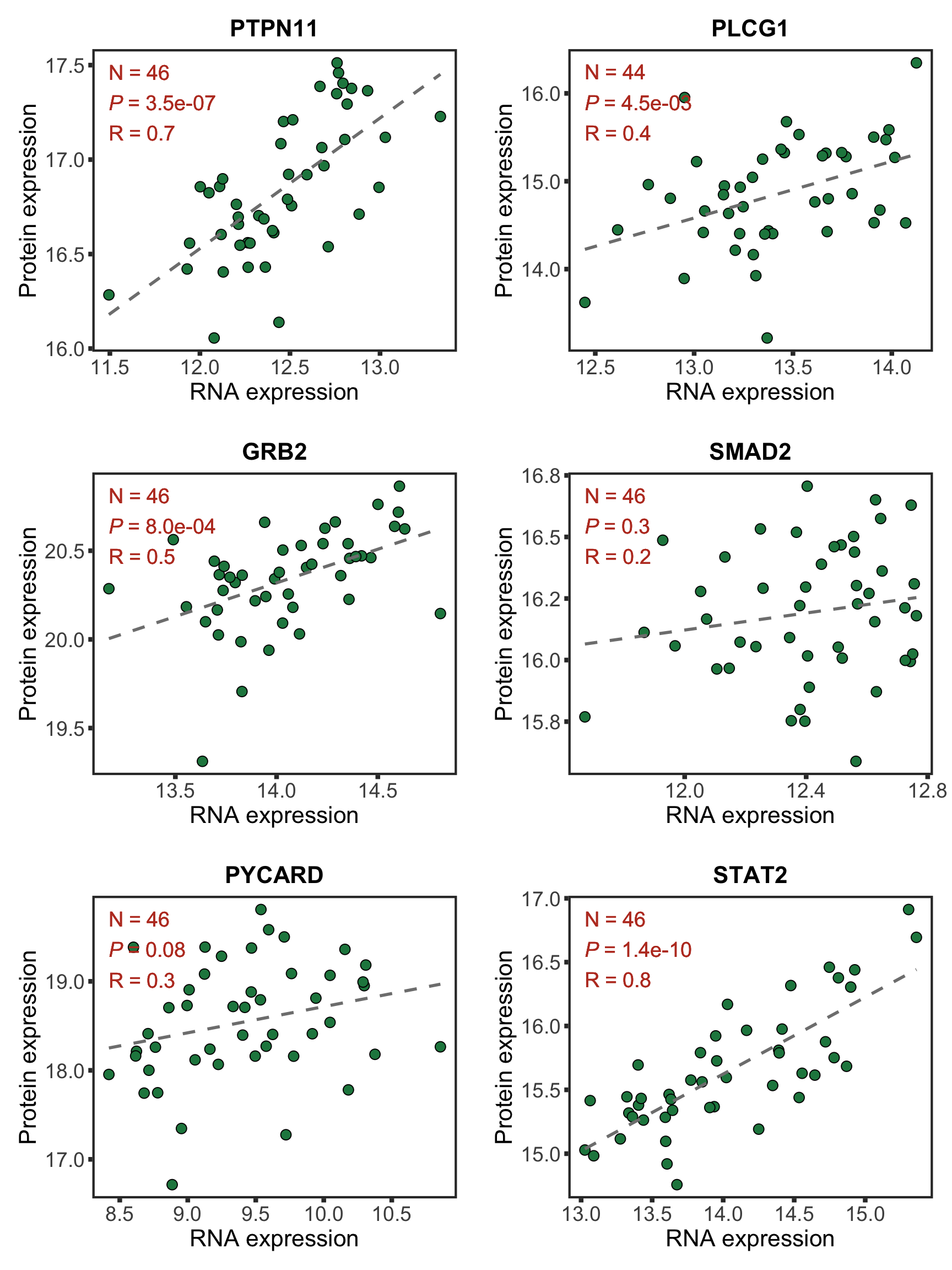
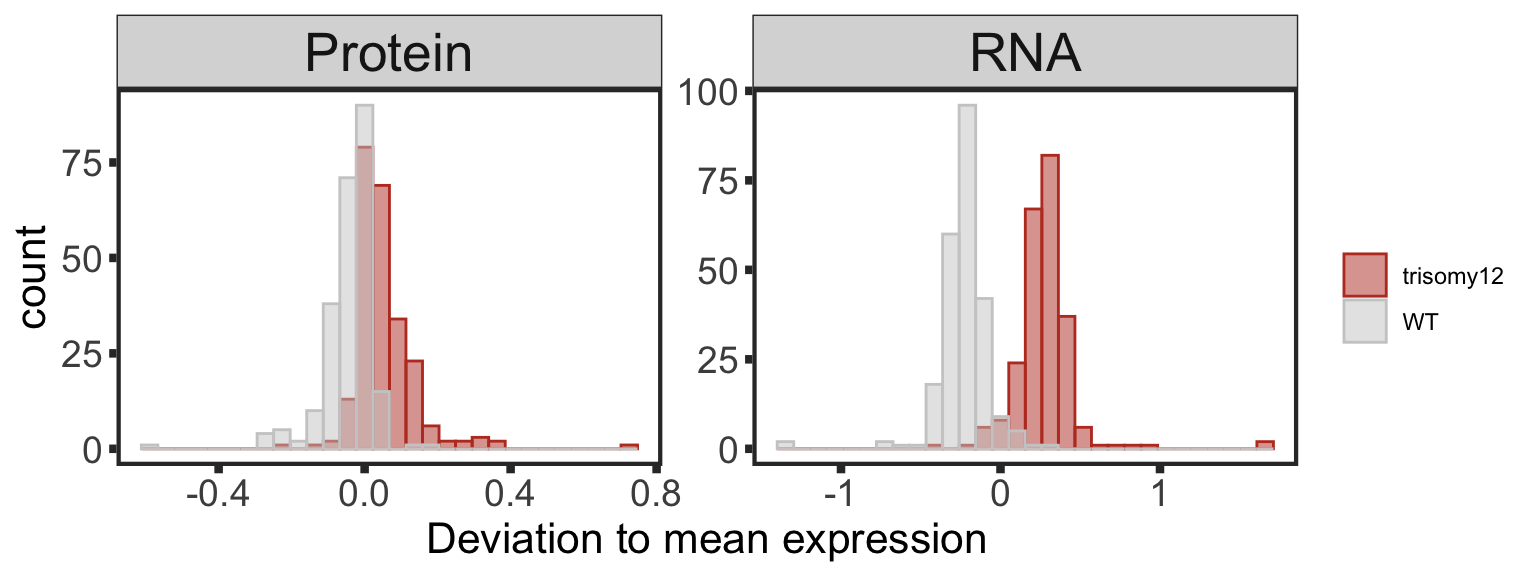
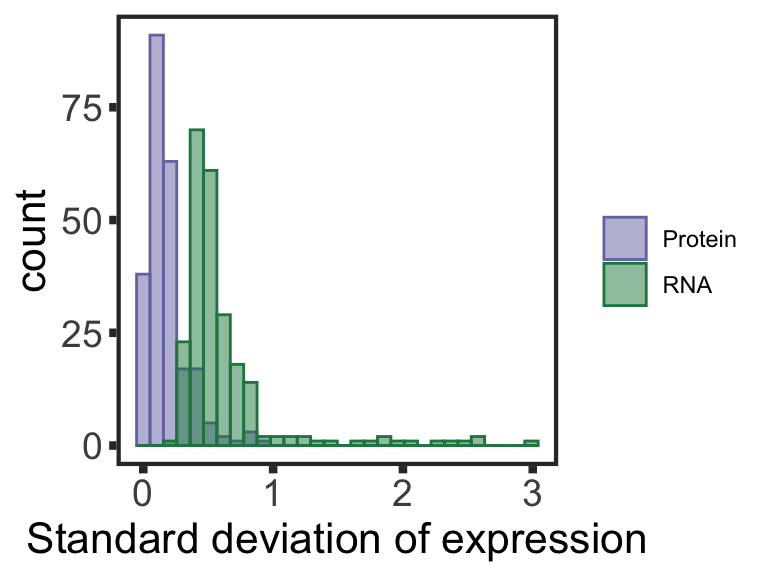
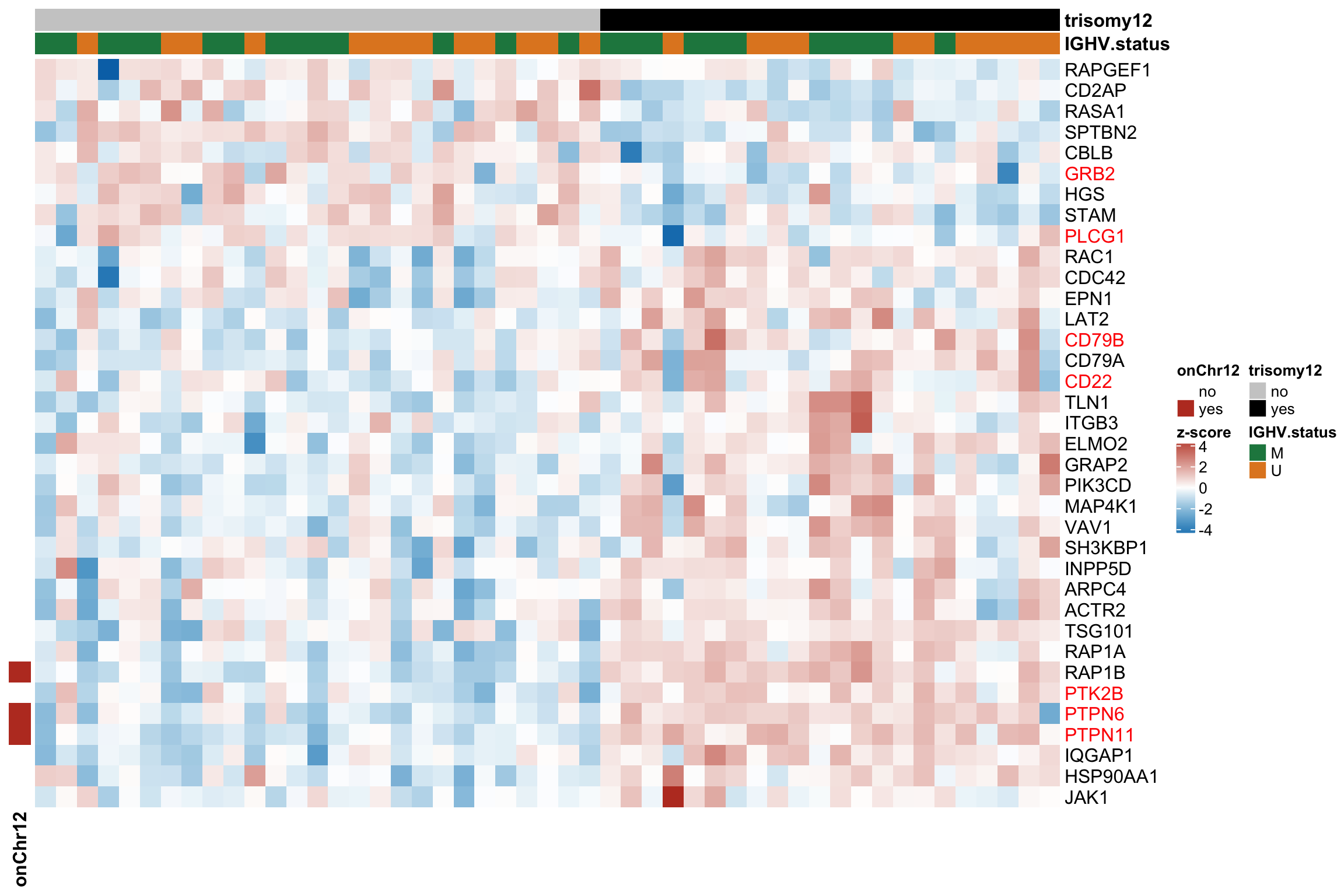
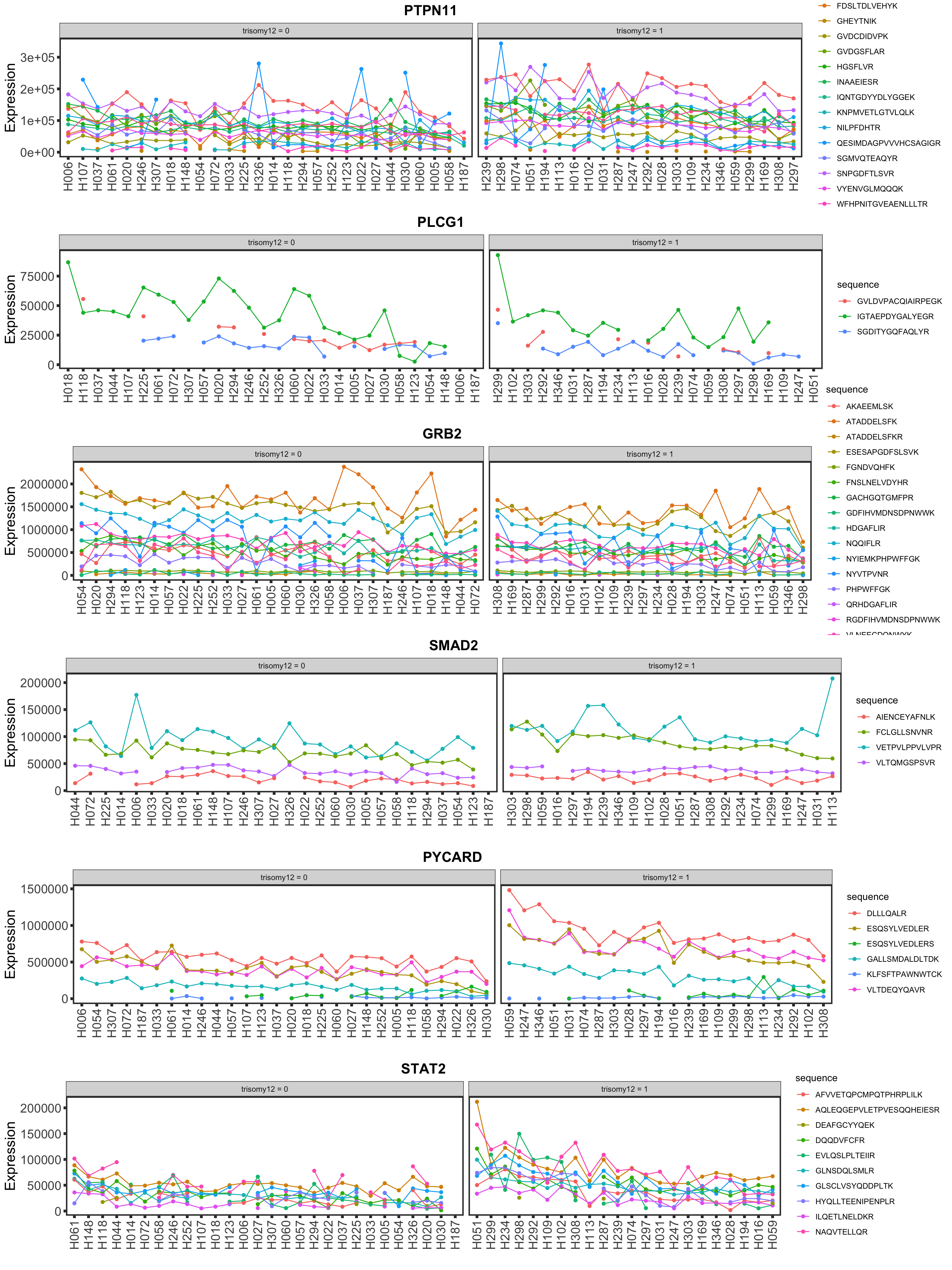
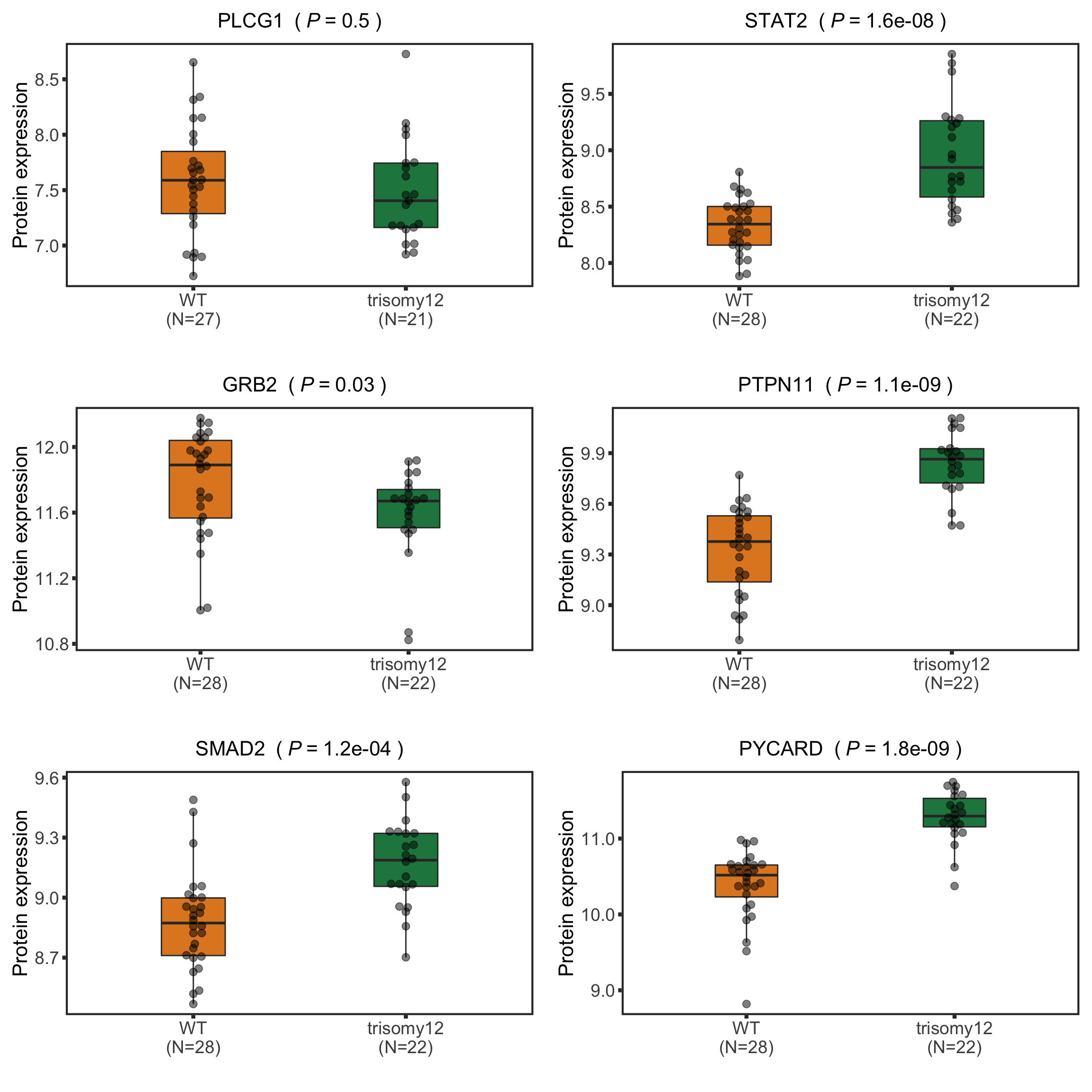 Now PLCG1 does not correlate with trisomy12, but PYCARD can be detected and correlate with trisomy12
Now PLCG1 does not correlate with trisomy12, but PYCARD can be detected and correlate with trisomy12