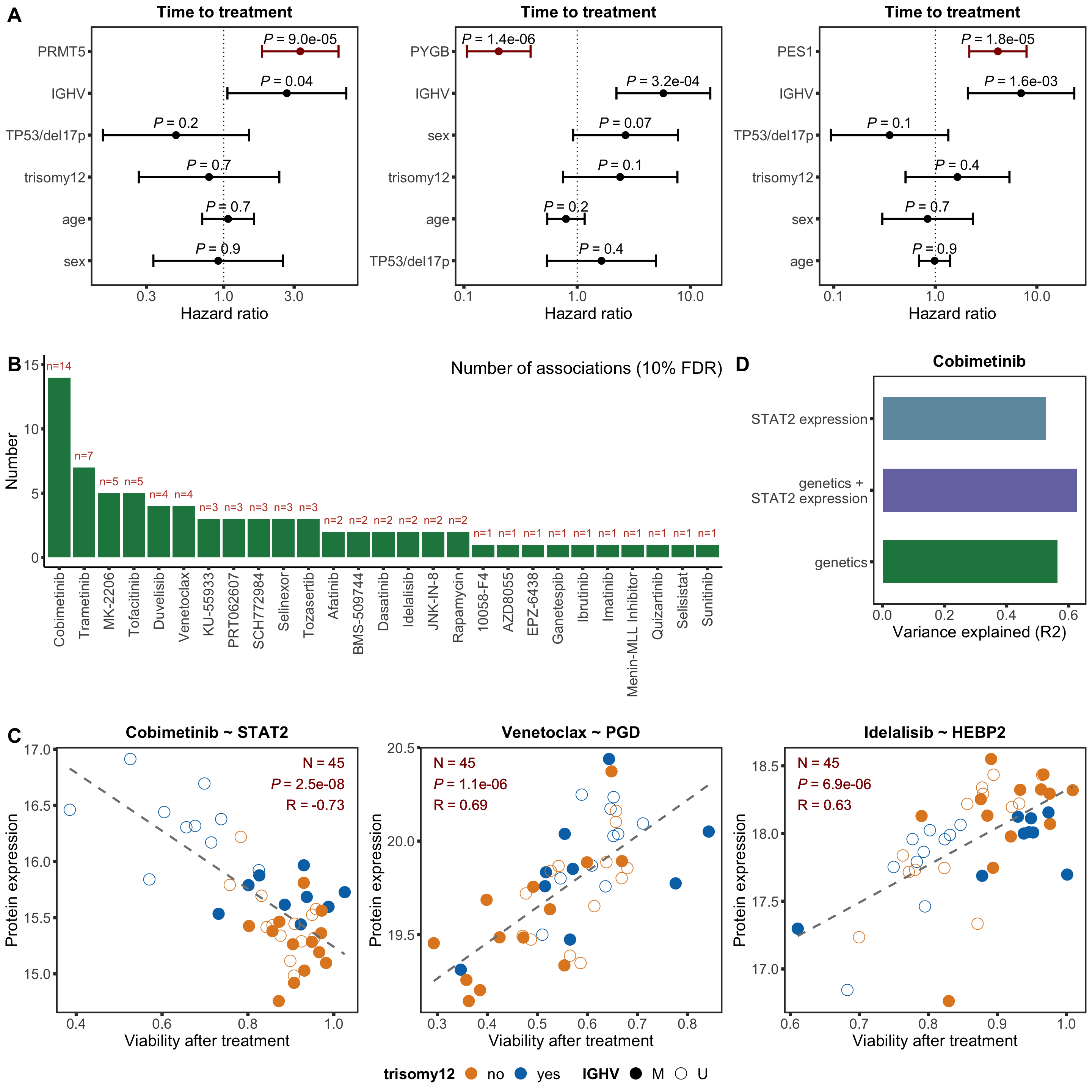
Correlation plot of selected candidates
plotDrugCorScatter <- function(inputTab, x, y, x_lab = "X", y_lab = "Y", title = "",
col = NULL, showR2 = TRUE, annoPos = "right",
dotCol = colList, textCol="darkred") {
#prepare table for plotting
plotTab <- tibble(x = inputTab[[x]],y=inputTab[[y]])
if (!is.null(col)) plotTab <- mutate(plotTab, status = inputTab[[col]])
plotTab <- filter(plotTab, !is.na(x), !is.na(y))
#prepare annotation values
corRes <- cor.test(plotTab$x, plotTab$y)
pval <- formatNum(corRes$p.value, digits = 1, format = "e")
Rval <- formatNum(corRes$estimate, digits = 1, format = "e")
R2val <- formatNum(corRes$estimate^2, digits = 1, format = "e")
Nval <- nrow(plotTab)
annoP <- bquote(italic("P")~"="~.(pval))
if (showR2) {
annoCoef <- bquote(R^2~"="~.(R2val))
} else {
annoCoef <- bquote(R~"="~.(Rval))
}
annoN <- bquote(N~"="~.(Nval))
corPlot <- ggplot(plotTab, aes(x = x, y = y))
if (!is.null(col)) {
corPlot <- corPlot + geom_point(aes(fill = status), shape =21, size =3) +
scale_fill_manual(values = dotCol)
} else {
corPlot <- corPlot + geom_point(fill = dotCol[1], shape =21, size=3)
}
corPlot <- corPlot + geom_smooth(formula = y~x,method = "lm", se=FALSE, color = "grey50", linetype ="dashed" )
if (annoPos == "right") {
corPlot <- corPlot + annotate("text", x = max(plotTab$x), y = Inf, label = annoN,
hjust=1, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$x), y = Inf, label = annoP,
hjust=1, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$x), y = Inf, label = annoCoef,
hjust=1, vjust =6, size = 5, parse = FALSE, col= textCol)
} else if (annoPos== "left") {
corPlot <- corPlot + annotate("text", x = min(plotTab$x), y = Inf, label = annoN,
hjust=0, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$x), y = Inf, label = annoP,
hjust=0, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$x), y = Inf, label = annoCoef,
hjust=0, vjust =6, size = 5, parse = FALSE, col= textCol)
}
corPlot <- corPlot + ylab(y_lab) + xlab(x_lab) + ggtitle(title) +
scale_y_continuous(labels = scales::number_format(accuracy = 0.1)) +
scale_x_continuous(labels = scales::number_format(accuracy = 0.1)) +
theme_full
corPlot
}
pairList <- list(c("Cobimetinib","STAT2"),c("Venetoclax","PGD"),c("Idelalisib", "HEBP2"))
plotList <- lapply(pairList, function(pair) {
textCol <- "darkred"
drugName <- pair[1]
proteinName <- pair[2]
id <- rownames(protCLL)[match(proteinName, rowData(protCLL)$hgnc_symbol)]
plotTab <- tibble(patID = colnames(viabMat),
viab = viabMat[drugName,],
expr = proMat[id,]) %>%
mutate(IGHV = protCLL[,patID]$IGHV.status,
trisomy12 = protCLL[,patID]$trisomy12) %>%
mutate(trisomy12 = ifelse(trisomy12 ==1,"yes","no")) %>%
filter(!is.na(viab),!is.na(expr))
pval <- formatNum(filter(resTab.sig, Drug == drugName, symbol == proteinName)$P.Value, digits = 1, format = "e")
Rval <- sprintf("%1.2f",cor(plotTab$viab, plotTab$expr))
Nval <- nrow(plotTab)
annoP <- bquote(italic("P")~"="~.(pval))
annoN <- bquote(N~"="~.(Nval))
annoCoef <- bquote(R~"="~.(Rval))
corPlot <- ggplot(plotTab, aes(x = viab, y = expr)) +
geom_point(aes(col = trisomy12, shape = IGHV), size=5) +
scale_shape_manual(values = c(M = 19, U = 1)) +
scale_color_manual(values = c(yes = colList[2], no = colList[3])) +
geom_smooth(formula = y~x,method = "lm", se=FALSE, color = "grey50", linetype ="dashed" ) +
ggtitle(sprintf("%s ~ %s", drugName, proteinName)) +
ylab("Protein expression") + xlab("Viability after treatment") +
theme_full +
theme(legend.position = "bottom",
legend.text = element_text(size=15), legend.title = element_text(size=15, face = "bold"))
if (Rval < 0) annoPos <- "right" else annoPos <- "left"
if (annoPos == "right") {
corPlot <- corPlot + annotate("text", x = max(plotTab$viab), y = Inf, label = annoN,
hjust=1, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$viab), y = Inf, label = annoP,
hjust=1, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = max(plotTab$viab), y = Inf, label = annoCoef,
hjust=1, vjust =6, size = 5, parse = FALSE, col= textCol)
} else if (annoPos== "left") {
corPlot <- corPlot + annotate("text", x = min(plotTab$viab), y = Inf, label = annoN,
hjust=0, vjust =2, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$viab), y = Inf, label = annoP,
hjust=0, vjust =4, size = 5, parse = FALSE, col= textCol) +
annotate("text", x = min(plotTab$viab), y = Inf, label = annoCoef,
hjust=0, vjust =6, size = 5, parse = FALSE, col= textCol)
}
corPlot <- corPlot + ylab("Protein expression") + xlab("Viability after treatment") +
scale_y_continuous(labels = scales::number_format(accuracy = 0.1)) +
scale_x_continuous(labels = scales::number_format(accuracy = 0.1))
corPlot
})
legend <- cowplot::get_legend(plotList[[1]])
plotNoLegend <- lapply(plotList, function(p) p + theme(legend.position = "none"))
drugCor <- plot_grid(plot_grid(plotlist = plotNoLegend, ncol =3),legend,ncol=1, rel_heights = c(0.9,0.1))
drugCor

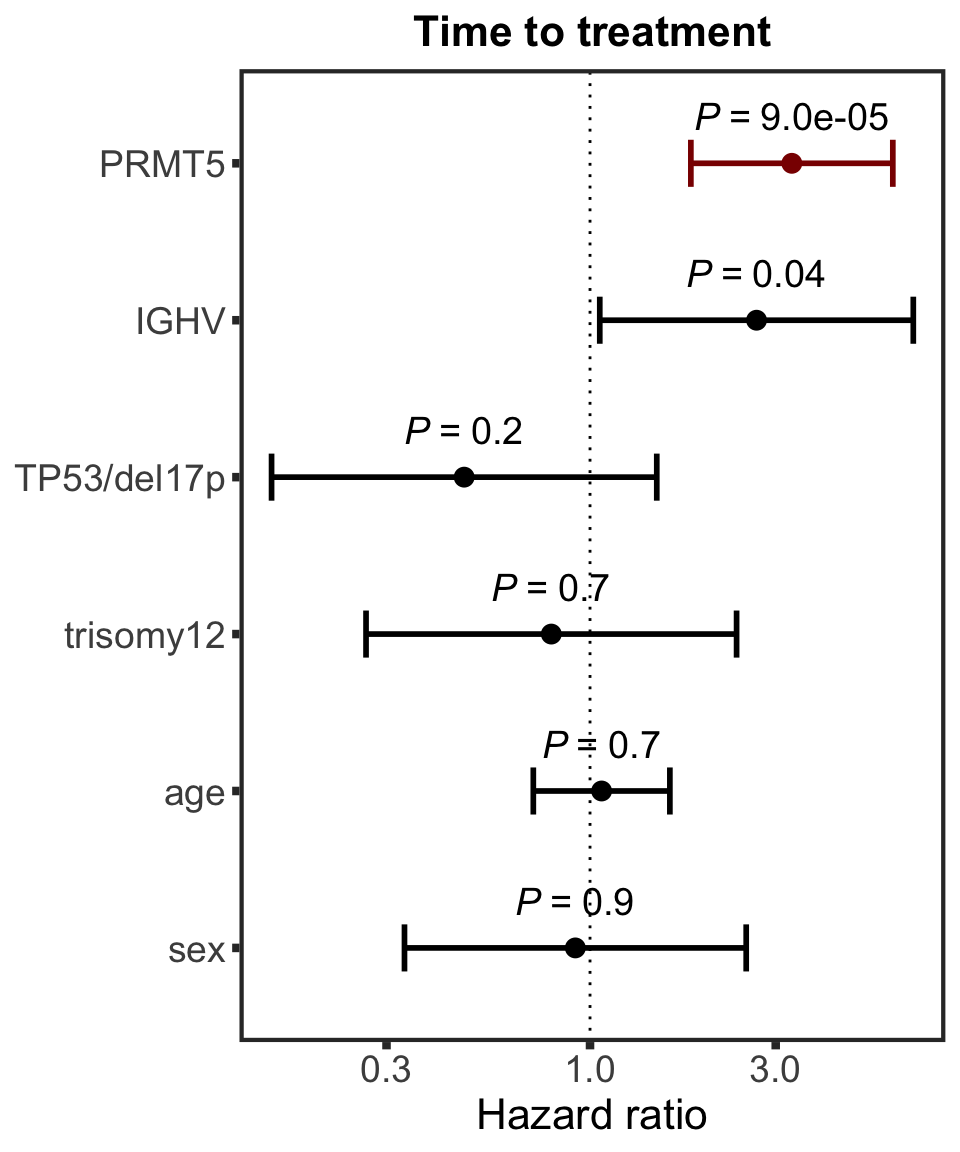
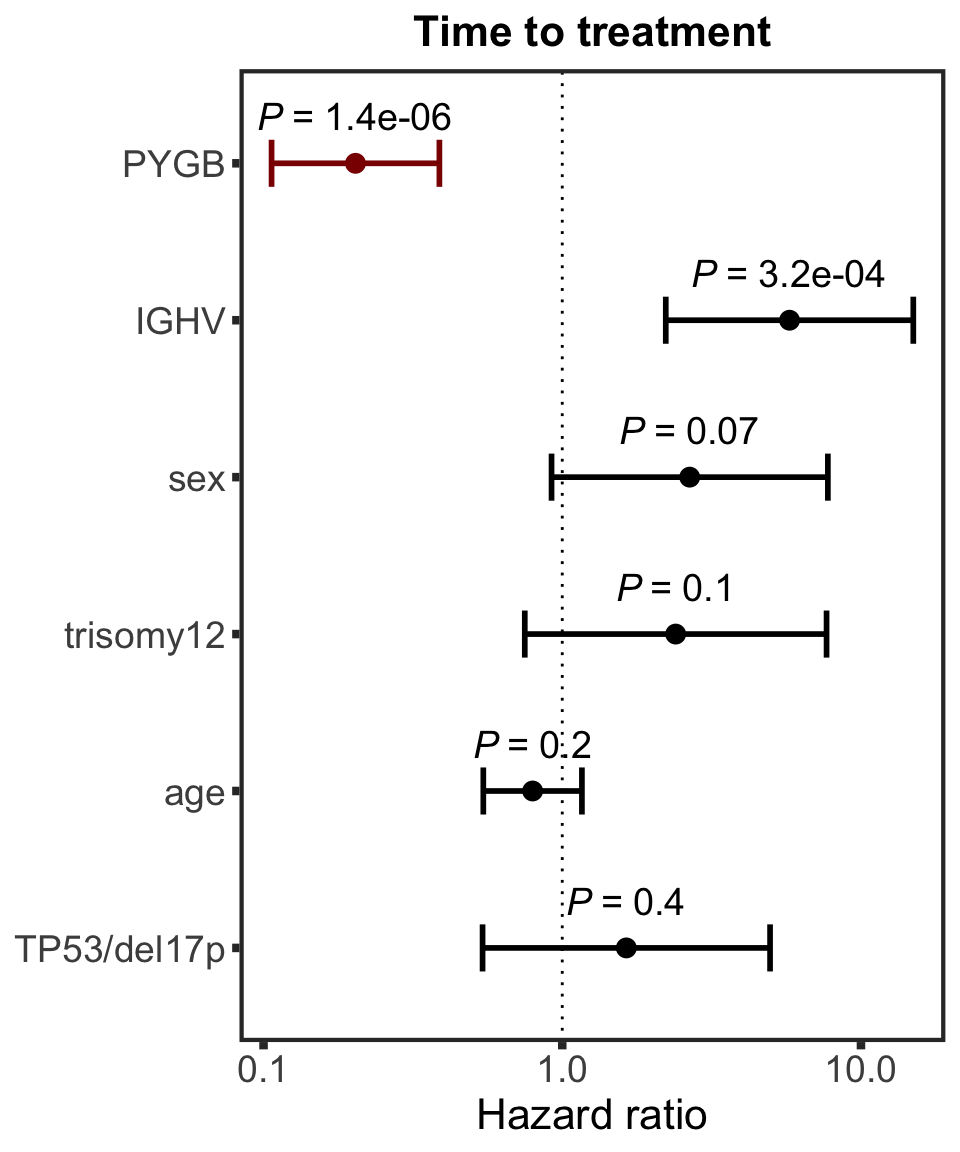
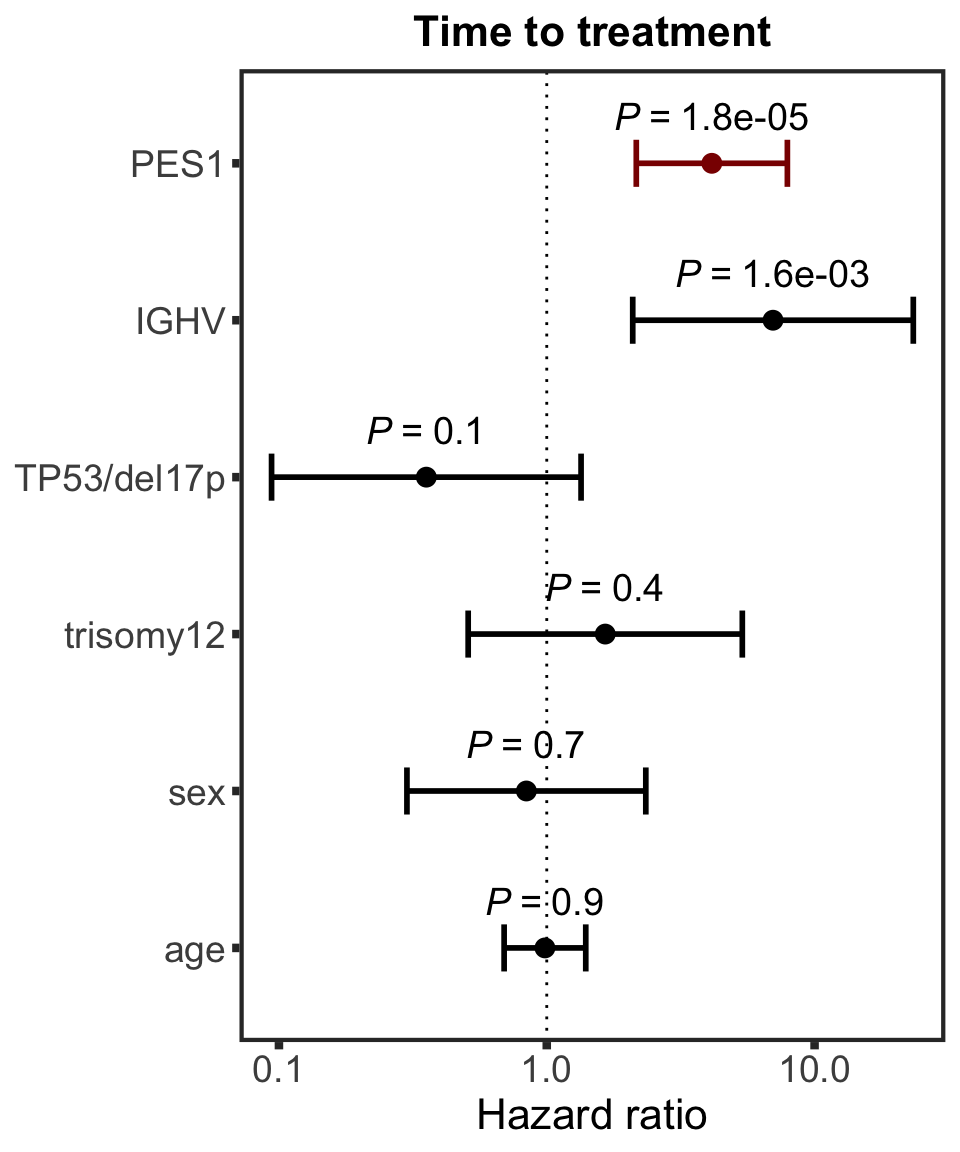
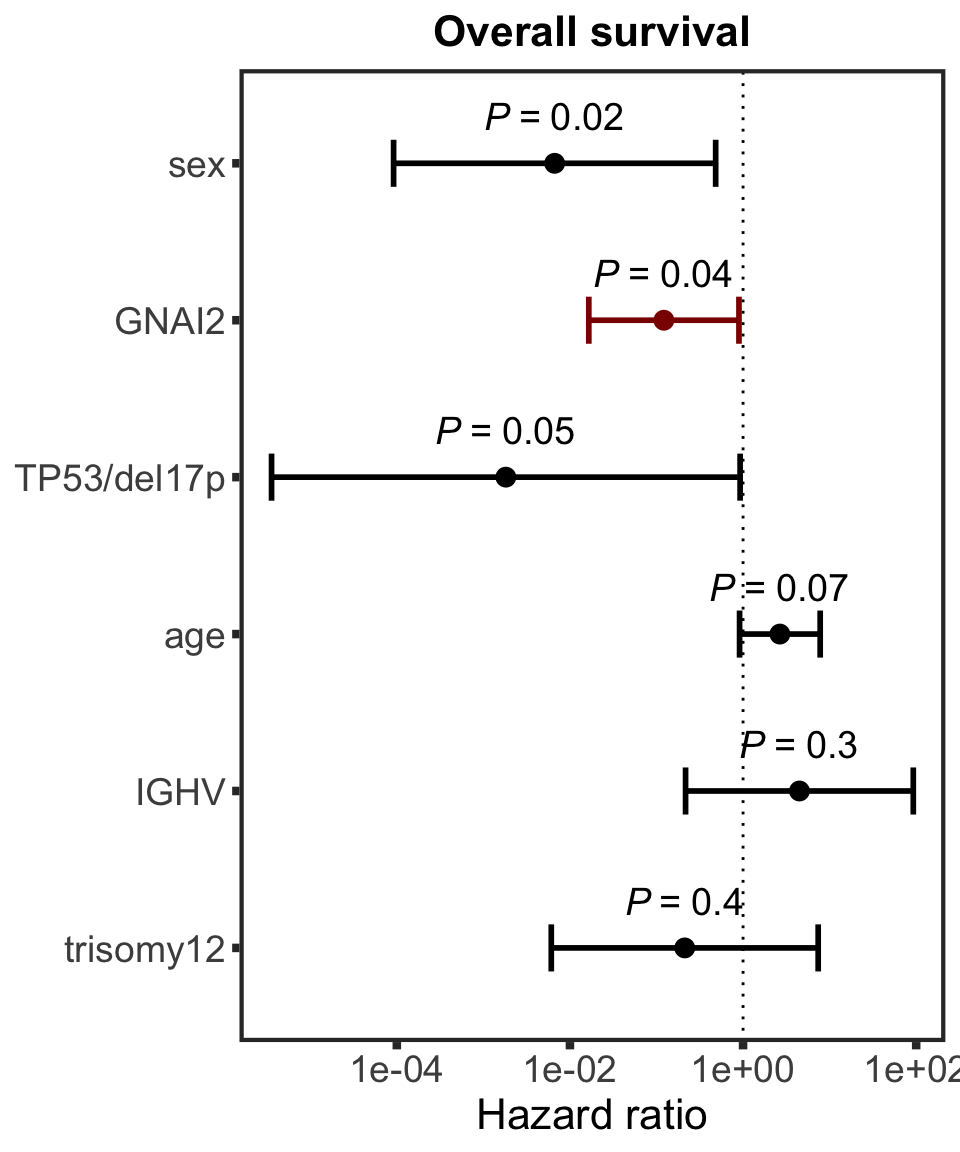
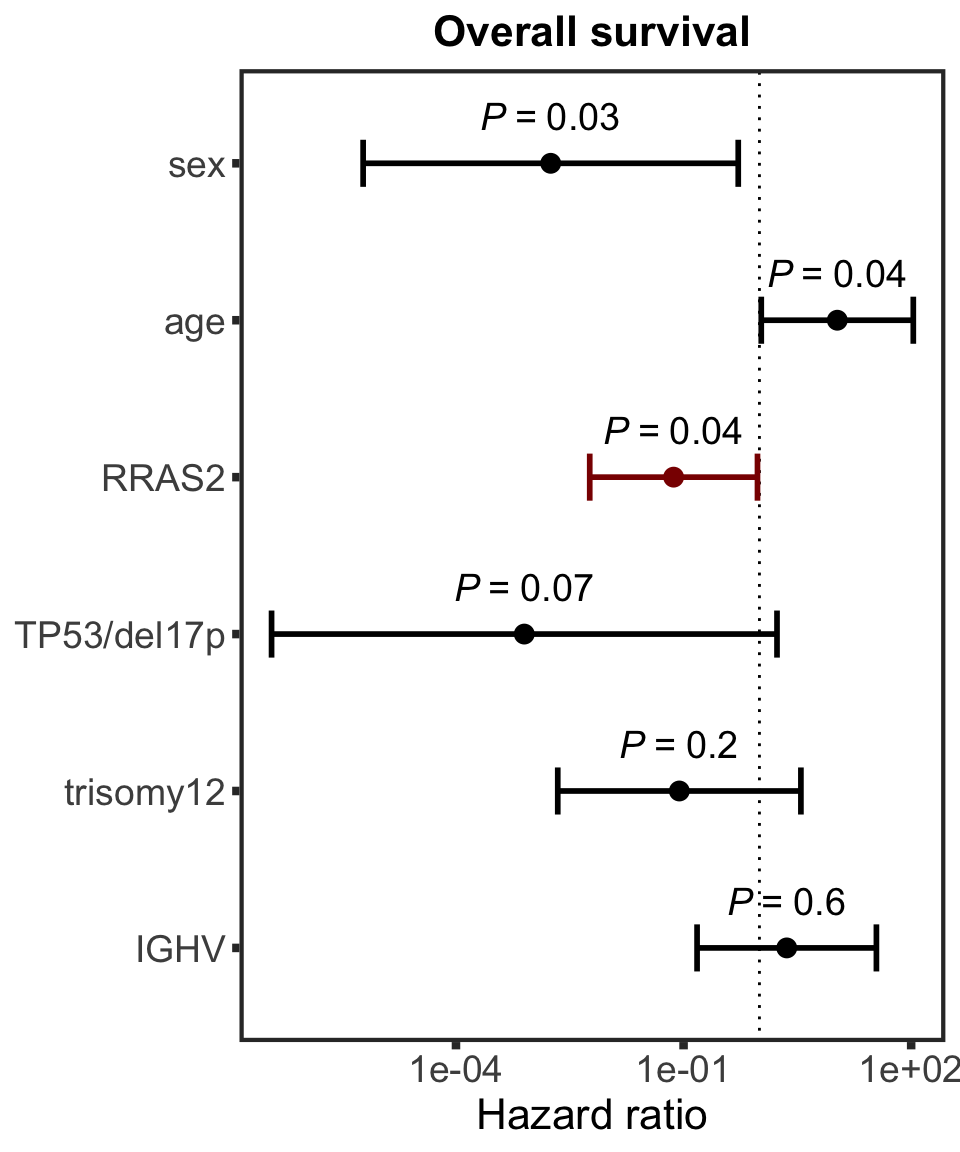
Association test with blocking for IGHV and trisomy12
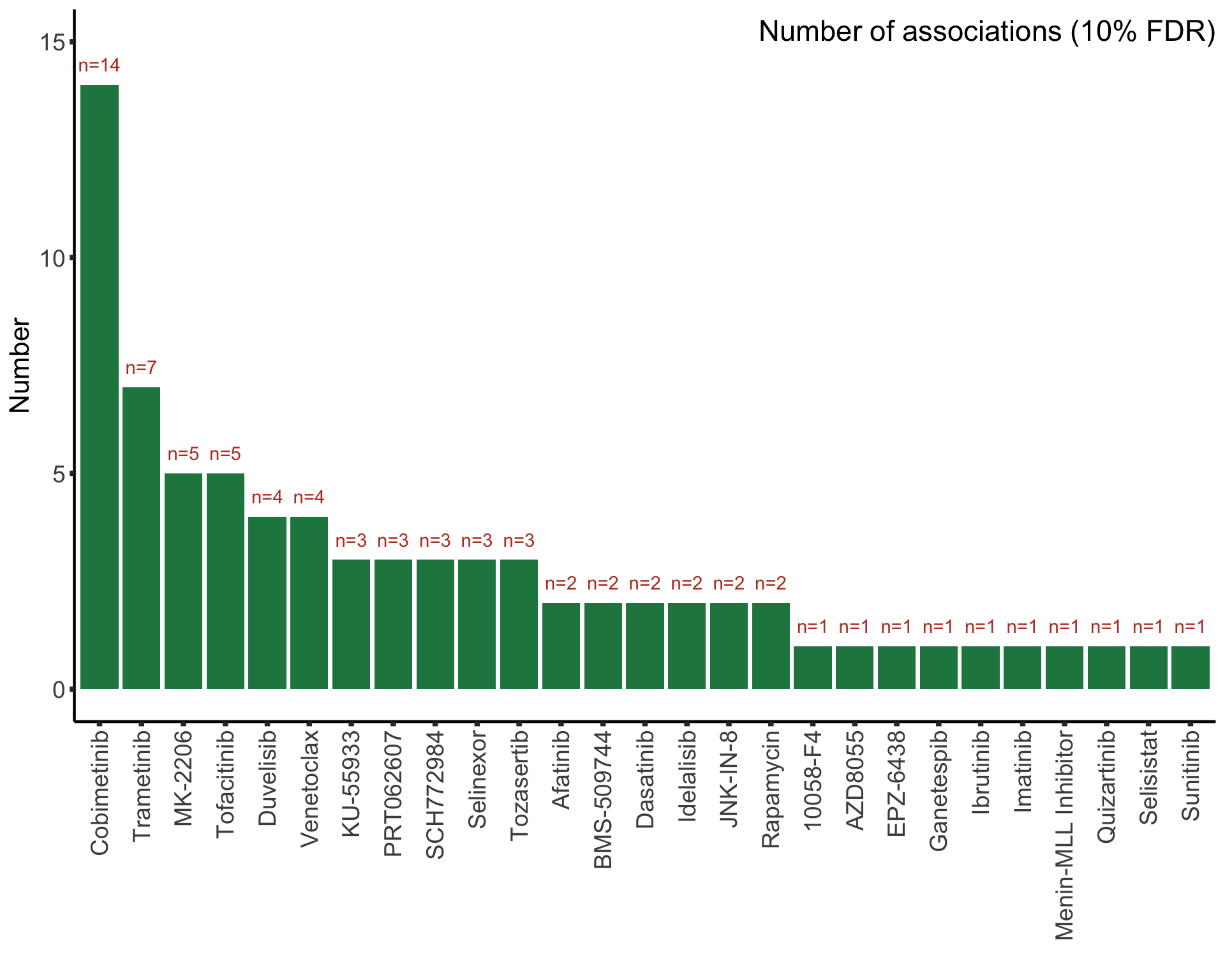
testList <- filter(resTab.auc, adj.P.Val < 0.1)
resTab.auc.block <- lapply(seq(nrow(testList)),function(i) {
pair <- testList[i,]
expr <- proMat[pair$id,]
viab <- viabMat[pair$Drug, ]
ighv <- protCLL[,colnames(viabMat)]$IGHV.status
tri12 <- protCLL[,colnames(viabMat)]$trisomy12
res <- anova(lm(viab~ighv+tri12+expr))
data.frame(id = pair$id, P.Value = res["expr",]$`Pr(>F)`, symbol = pair$symbol,
Drug = pair$Drug,
P.Value.IGHV = res["ighv",]$`Pr(>F)`,P.Value.trisomy12 = res["tri12",]$`Pr(>F)`,
P.Value.noBlock = pair$P.Value,
stringsAsFactors = FALSE)
}) %>% bind_rows() %>% mutate(adj.P.Val = p.adjust(P.Value, method = "BH")) %>% arrange(P.Value)
Assocations that are still significant
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()
The above mentioned pairs are still significant.
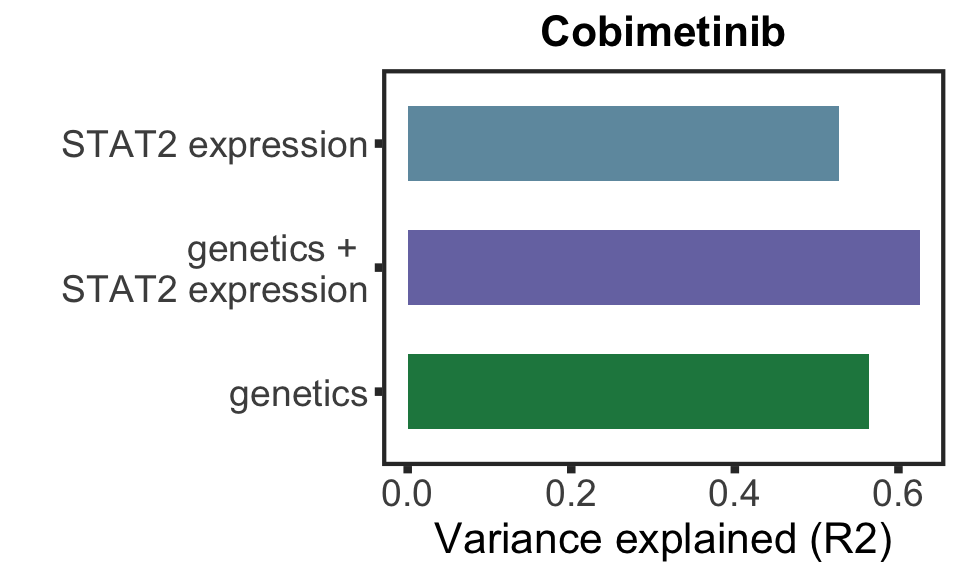
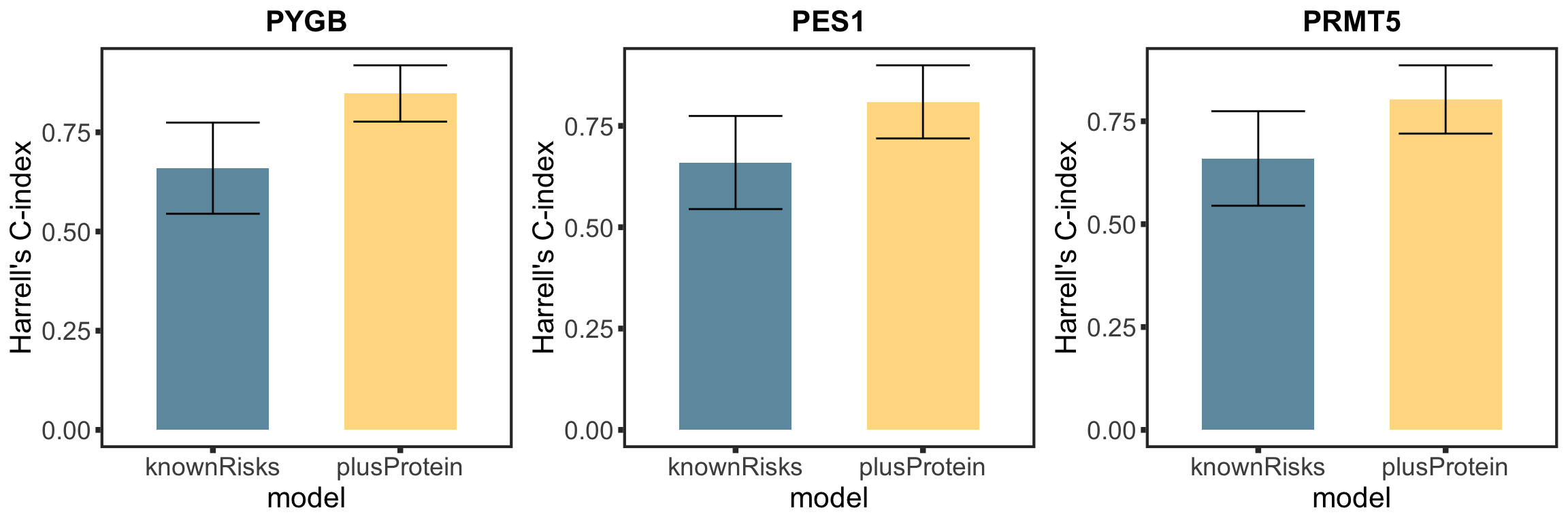
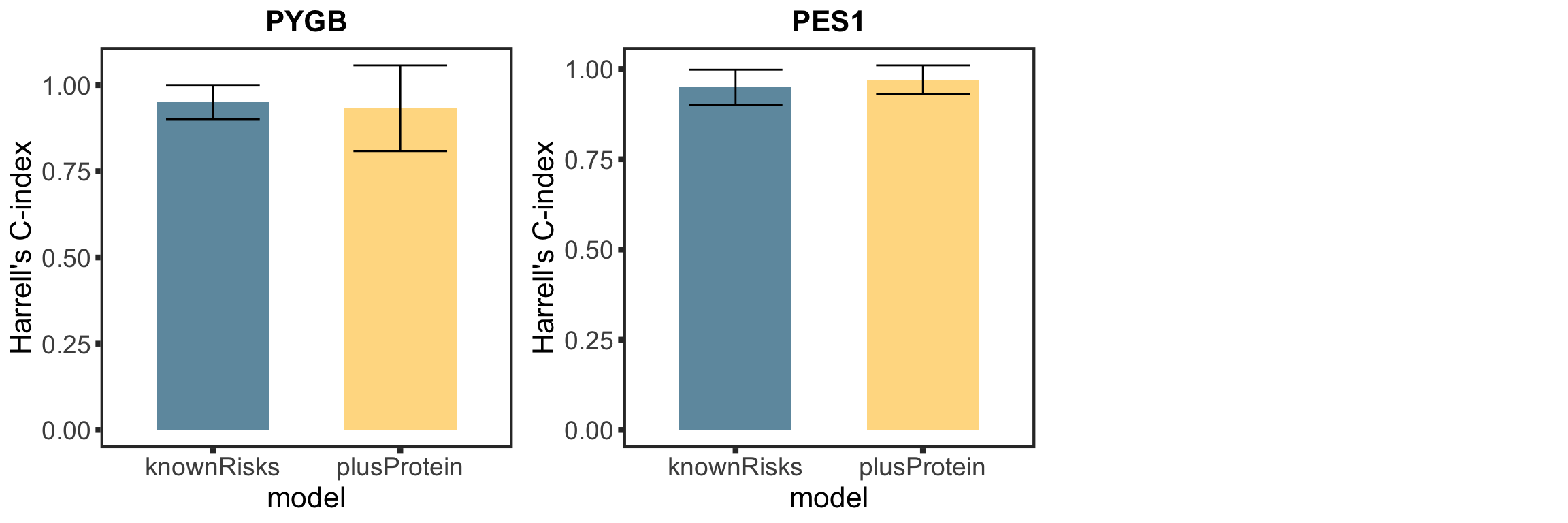
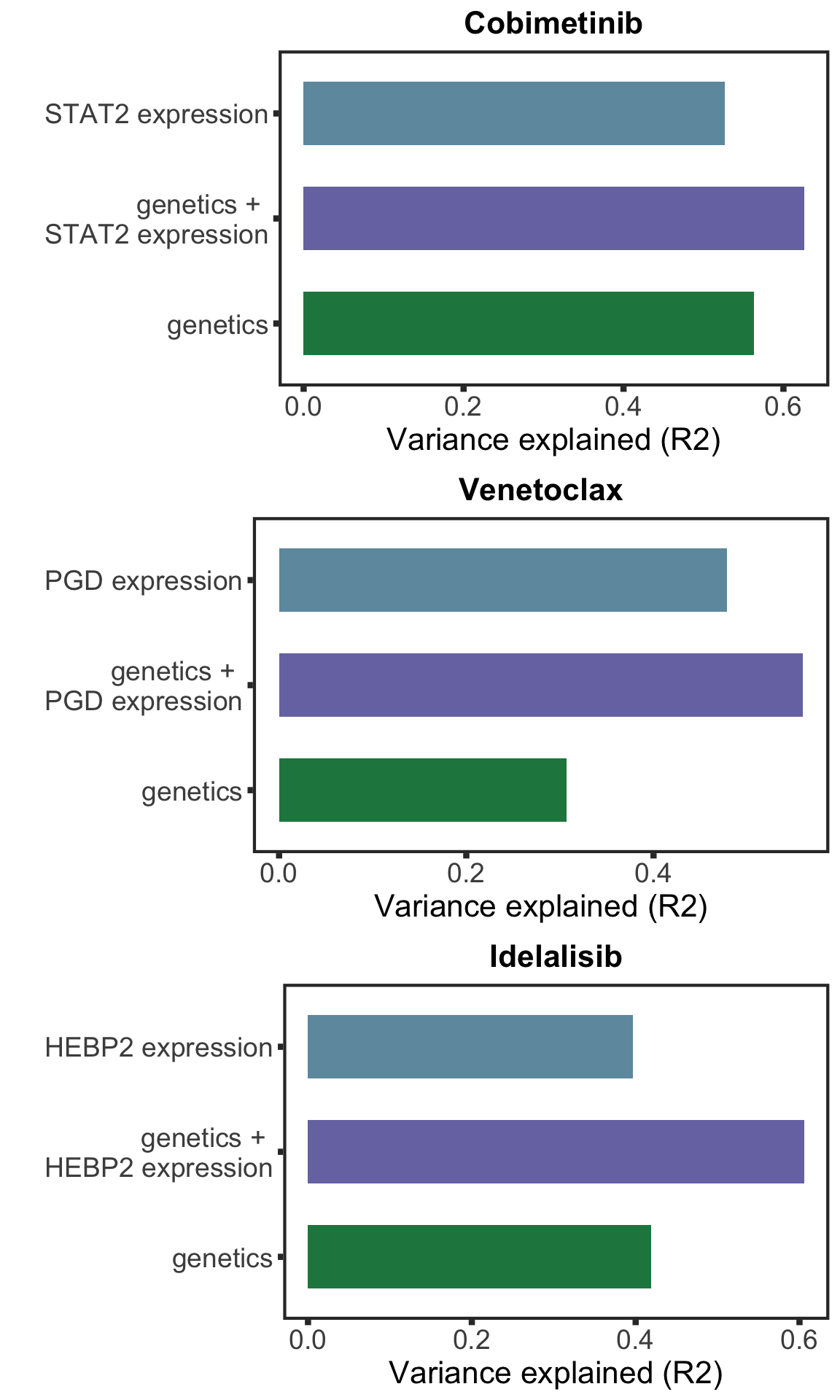 Based on the plot of R2 values, including protein expression in multi-variate model could explain additional variance in drug responses compared to genetic alone. For Venetoclax, using a single protein expression value of PGD already explain more variance than genetics.
Based on the plot of R2 values, including protein expression in multi-variate model could explain additional variance in drug responses compared to genetic alone. For Venetoclax, using a single protein expression value of PGD already explain more variance than genetics.