Plot RNA and protein expression on genomic coordinates overlayed with copy number changes
Junyan Lu
2020-02-27
Last updated: 2020-05-25
Checks: 6 1
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/complexAnalysis_IGHV_cache/
Ignored: analysis/complexAnalysis_trisomy12_alteredPQR_cache/
Ignored: analysis/complexAnalysis_trisomy12_cache/
Ignored: analysis/correlateCLLPD_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/complexAnalysis_IGHV.Rmd
Untracked: analysis/complexAnalysis_trisomy12.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/default.css
Untracked: analysis/del11q.pdf
Untracked: analysis/del11q_norm.pdf
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/plotCNV_del11q.pdf
Untracked: analysis/plotExpressionCNV.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/style.css
Untracked: analysis/trisomy12.pdf
Untracked: analysis/trisomy12_norm.pdf
Untracked: code/AlteredPQR.R
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/example_proteomics_data
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/proteins_in_complexes
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/LUMOS_processed.RData
Untracked: output/cnv_plots.zip
Untracked: output/cnv_plots/
Untracked: output/cnv_plots_norm.zip
Untracked: output/dxdCLL.RData
Untracked: output/exprCNV.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/plotCNV_allChr11_diff.pdf
Untracked: output/plotCNV_del11q_sum.pdf
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_LUMOS_20200430.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/splicingResults.RData
Untracked: output/timsTOF_processed.RData
Untracked: plotCNV_del11q_diff.pdf
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateCLLPD.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
load("../../var/ddsrna_180717.RData")
load("../../var/patmeta_200424.RData")
load("../../var/proteomic_LUMOS_20200430.RData")
load("../../var/CNV_onChrom.RData")Annotated mRNAs with genomic coordinate
mart<-useMart(biomart="ENSEMBL_MART_ENSEMBL",
dataset="hsapiens_gene_ensembl",
host="grch37.ensembl.org")
locAnno <- getBM(values = rownames(dds) ,mart = mart,
attributes = c("ensembl_gene_id","start_position","end_position","transcript_length","cds_length"),
filters = "ensembl_gene_id")
lengthTab <- group_by(locAnno, ensembl_gene_id) %>%
summarise(avgTrLen = median(transcript_length,na.rm=TRUE),
avgCDSLen = median(cds_length, na.rm=TRUE))
geneAnno <- distinct(locAnno, ensembl_gene_id, .keep_all = TRUE) %>%
select(-transcript_length,-cds_length) %>%
left_join(lengthTab, by = "ensembl_gene_id") %>%
data.frame(stringsAsFactors = FALSE) %>%
remove_rownames() %>%
column_to_rownames("ensembl_gene_id")newAnno <- cbind(rowData(dds),geneAnno)
rowData(dds) <- newAnno
dds <- dds[rowData(dds)$chromosome %in% c(as.character(seq(22)),"X","Y"),]
dds <- dds[!rowData(dds)$symbol %in% c("",NA),]Prepare protein expression and RNA expression data
#overSample <- intersect(colnames(dds),colnames(protCLL))
ddsSub <- dds[, colnames(dds) %in% colnames(protCLL)]
protSub <- protCLL[rowData(protCLL)$ensembl_gene_id %in% rownames(ddsSub)]Generate an assay experiment object
glog2 <- function(x) ((asinh(x)-log(2))/log(2))
rnaMat <- counts(ddsSub, normalized = TRUE)
#rnaMat <- glog2(rnaMat) + 1
rnaTab <- data.frame(rnaMat) %>% rownames_to_column("id") %>%
mutate(len = rowData(dds[id,])$avgTrLen) %>%
gather(key = "patID", value = "count",-id,-len) %>%
mutate(expr = glog2((count/len)*1000) +1) %>% #normalize by length
select(-len,-count)
protMat <- assays(protSub)[["count"]]
protMat <- proDA::median_normalization(protMat)
rownames(protMat)<-rowData(protSub)$ensembl_gene_id
protTab <- data.frame(protMat) %>% rownames_to_column("id") %>%
mutate(len = rowData(dds)[match(id,rownames(dds)),]$avgCDSLen) %>%
filter(!is.na(len)) %>%
gather(key = "patID", value = "count",-id,-len) %>%
mutate(expr = (count/len)*200) %>% #normalize by length
select(-len,-count)
#exprTab <- left_join(rnaTab, protTab, by = c("id","patID"))
annoTab <- rowData(ddsSub) %>% data.frame() %>%
rownames_to_column("id") %>%
mutate(chr = as.numeric(chromosome)) %>%
mutate(ChromID = ifelse(!is.na(chr),sprintf("chr%02s",chr),sprintf("chr%s",chromosome))) %>%
select(id,symbol,ChromID, start_position, end_position)
rnaTab <- left_join(rnaTab, annoTab, by = "id") %>%
as_tibble() %>% mutate_if(is.character, as.factor)
protTab <- left_join(protTab, annoTab, by = "id") %>%
as_tibble() %>% filter(!is.na(ChromID), !is.na(expr)) %>%
mutate_if(is.character, as.factor)allBand <- cytoBand %>%
mutate(chromStart = chromStart/10^6,
chromEnd = chromEnd/10^6,
chromMid = chromMid/10^6) %>%
dplyr::rename(band = bandname)
allLine <- lineTab %>%
mutate(SegmentMean = case_when(
SegmentMean > 2 ~ 2,
SegmentMean < -2 ~ -2,
TRUE ~ SegmentMean
)) %>%
mutate(Start = Start/10^6, End = End/10^6)
allProtTab <- protTab %>%
mutate(start_position = start_position/10^6,
end_position = end_position/10^6,
mid_position = (start_position + end_position)/2)
allRnaTab <- rnaTab %>%
mutate(start_position = start_position/10^6,
end_position = end_position/10^6,
mid_position = (start_position + end_position)/2)
save(allBand, allLine, allProtTab, allRnaTab,
file = "../output/exprCNV.RData")load("../output/exprCNV.RData")Normalize protein and RNA expression
normalized <- TRUE
#if perform normalization
if (normalized) {
#for protein
exprMat <- select(allProtTab,patID, id,expr) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allProtTab <- select(allProtTab, -expr) %>% left_join(normTab, by = c("patID","id"))
#for RNA
exprMat <- select(allRnaTab,patID, id,expr) %>%
spread(key = patID, value =expr) %>% data.frame() %>%
column_to_rownames("id") %>% as.matrix()
qm <- jyluMisc::mscale(exprMat, useMad = F)
normTab <- data.frame(qm) %>% rownames_to_column("id") %>%
gather(key = "patID", value = "expr", -id)
allRnaTab <- select(allRnaTab, -expr) %>% left_join(normTab, by = c("patID","id"))
}Warning: replacing previous import 'cowplot::ggsave' by 'ggplot2::ggsave'
when loading 'jyluMisc'Registered S3 method overwritten by 'sets':
method from
print.element ggplot2Warning: Column `patID` joining factor and character vector, coercing into
character vectorWarning: Column `id` joining factor and character vector, coercing into
character vectorWarning: Column `patID` joining factor and character vector, coercing into
character vectorWarning: Column `id` joining factor and character vector, coercing into
character vectorFunction for plotting
plotExprCNV <- function(pat, chr, allBand, allLine, allProtTab, allRnaTab, ifTrend = FALSE,
startPos = -Inf, endPos= Inf, showLabel = "none", plotDiff = FALSE) {
multiPat <- length(unique(pat)) > 1
#table for cyto band
bandTab <- filter(allBand, ChromID == chr)
#table for expression
plotProtTab <- filter(allProtTab, ChromID == chr, patID %in% pat) %>%
mutate(expression = "protein") %>%
mutate_if(is.factor,as.character)
plotRnaTab <- filter(allRnaTab, ChromID == chr, patID %in% pat) %>%
mutate(expression = "rna") %>% mutate_if(is.factor,as.character)
if (!plotDiff) {
plotExprTab <- bind_rows(plotRnaTab, plotProtTab) %>%
filter(start_position > startPos, end_position < endPos)
} else {
plotProtTab <- plotProtTab %>% dplyr::rename(protein = expr)
plotRnaTab <- plotRnaTab %>% select(id, expr) %>%
dplyr::rename(rna = expr)
plotExprTab <- left_join(plotProtTab, plotRnaTab, by = "id") %>%
mutate(expr = protein-rna, expression = "protein-rna") %>%
filter(start_position > startPos, end_position < endPos) %>%
select(-protein,-rna)
}
if (multiPat) {
se <- function(x) sqrt(var(x,na.rm = T)/length(x))
plotExprTab <- group_by(plotExprTab, id, symbol, ChromID, start_position, end_position,mid_position, expression) %>%
summarise(upper = mean(expr,na.rm=T) + 1.96*se(expr), lower = mean(expr,na.rm=T) - 1.96*se(expr),
expr = mean(expr)) %>%
ungroup()
}
#table for copy number
plotLineTab <- filter(allLine, patID %in% pat, ChromID == chr)
#plot range
maxVal <- max(c(max(plotExprTab$expr,na.rm = T),max(plotLineTab$SegmentMean,na.rm = T)),na.rm = T) + 1
minVal <- min(c(min(plotExprTab$expr, na.rm = T),min(plotLineTab$SegmentMean,na.rm = T)),na.rm = T) - 1
#maxVal <- 5
#minVal <- -5
xMax <- max(bandTab$chromEnd, na.rm = T)
#main plot
gg <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal, ymax=maxVal,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal, hjust =1, angle = 90, size=2.5) +
geom_rect(data=plotLineTab,
mapping=aes(xmin=Start, xmax=End, ymin=SegmentMean,
ymax=SegmentMean+0.5,fill = set),alpha=0.2)
if (multiPat) {
gg <- gg + geom_errorbar(data = plotExprTab,
aes(x = mid_position, y = expr + 0.25, ymax = upper + 0.25, ymin=lower + 0.25),
col = "grey60")
}
gg <- gg + geom_rect(data = plotExprTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=expr, ymax=expr+0.5,
fill = expression, label = symbol), alpha =0.8) +
#scale_x_continuous(expand=c(0,0),limits = c(max(0,startPos),min(xMax,endPos))) +
scale_y_continuous(limits = c(minVal, maxVal), sec.axis = sec_axis(~./1, name = "Copy number")) +
coord_cartesian(xlim = c(max(0,startPos),min(xMax,endPos)), expand = FALSE)+
xlab("Genomic position [Mb]") +
ylab("Expression (normalized by length)") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
rna = "red", protein = "blue", `protein-rna` = "salmon",
WES = "darkgreen",WGS = "orange", Methylome = "purple")) +
scale_color_manual(values = c(protein = "blue",rna = "red",`protein-rna` = "salmon")) +
ggtitle(paste0(ifelse(multiPat,"all",pat),"_",chr)) +
theme(plot.title = element_text(face = "bold", size = 10, hjust = 0.3),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (showLabel != "none") {
gg <- gg +
ggrepel::geom_text_repel(data = filter(plotExprTab,
expression == showLabel),
aes(x=mid_position, y=expr, label = symbol))
}
if (ifTrend) {
gg <- gg + geom_smooth(data =filter(plotExprTab),
mapping = aes(y=expr, x= mid_position,
color = expression),
method = "loess", se=FALSE, span=0.2,
size =0.2)
}
#for legend
## if the patient has CNV data
lgTab <- tibble(x= seq(90),y=seq(90),
Expression = c(rep("protein",30), rep("rna",30),rep("protein-rna",30)),
CNV_data = rep(c("WES","WGS","Methylome"),30))
if (nrow(plotLineTab) >0) {
lgTab <- filter(lgTab, CNV_data %in% unique(plotLineTab$set),
Expression %in% unique(plotExprTab$expression))
lg <- ggplot(lgTab, aes(x=x,y=y)) +
geom_point(aes(fill = Expression), shape =22,size=3) +
geom_line(aes(color = CNV_data),size=5) +
scale_fill_manual(values = c(rna = "red", protein = "blue",`protein-rna` = "salmon")) +
scale_color_manual(values = c(WES = "darkgreen",WGS = "orange", Methylome = "purple")) +
theme(legend.position = "bottom")
} else {
lgTab <- filter(lgTab, Expression %in% unique(plotExprTab$expression))
lg <- ggplot(lgTab, aes(x=x,y=y)) +
geom_point(aes(fill = Expression), shape =22,size=3) +
scale_fill_manual(values = c(rna = "red", protein = "blue",`protein-rna` = "salmon")) +
theme(legend.position = "bottom")
}
lg <- get_legend(lg)
return(list(main=gg, legend = lg))
}Test of function using a example: chr11 of P0352
g <- plotExprCNV("P0352","chr11",allBand,allLine, allProtTab, allRnaTab, ifTrend = TRUE)Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelg$main`geom_smooth()` using formula 'y ~ x'Warning: Removed 236 rows containing non-finite values (stat_smooth).Warning: Removed 236 rows containing missing values (geom_rect).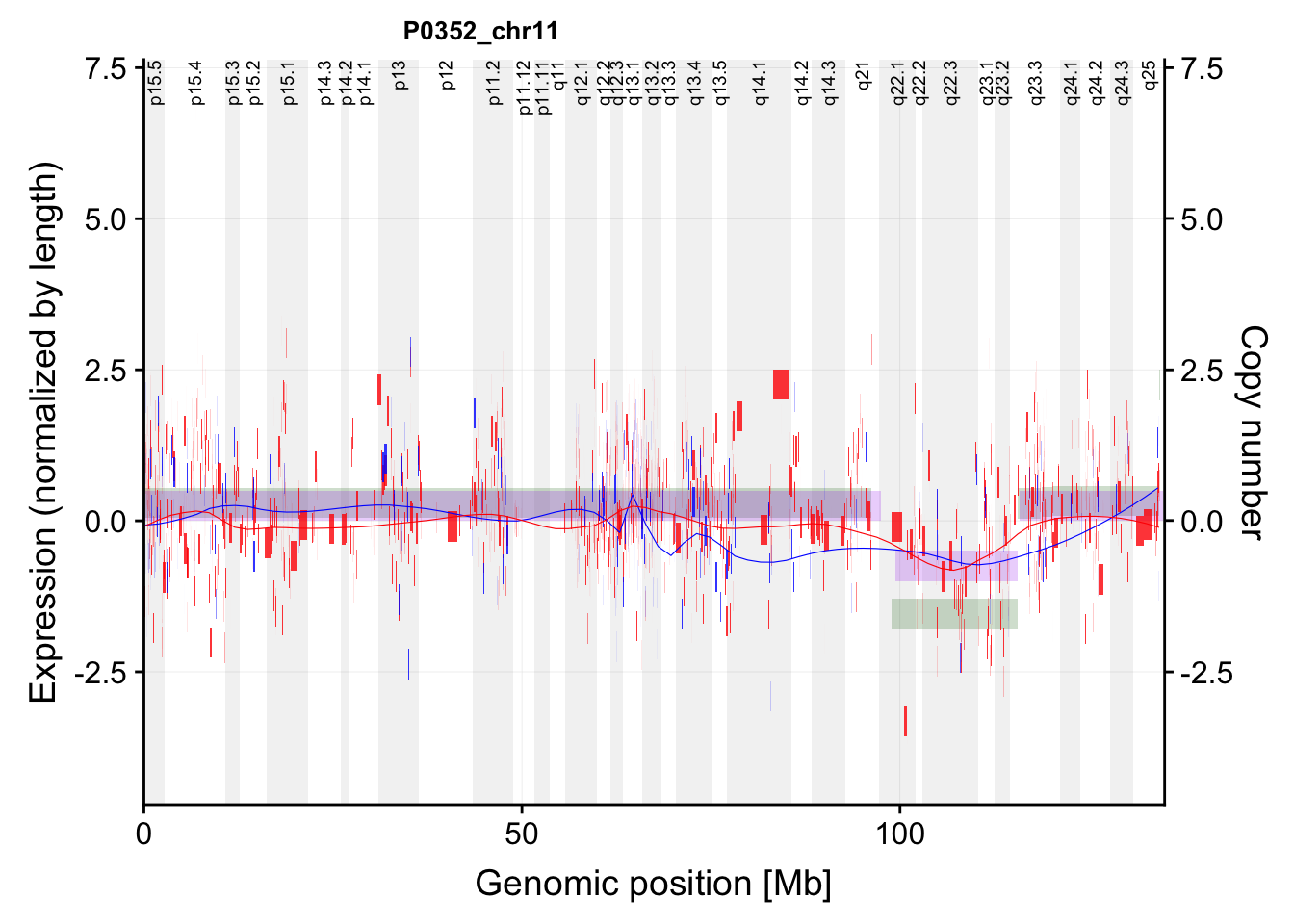
g <- plotExprCNV("P0352","chr11",allBand,allLine, allProtTab, allRnaTab, ifTrend = TRUE,
startPos = 97.2, endPos = 122.55, showLabel = "protein")Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelg$main`geom_smooth()` using formula 'y ~ x'Warning: Removed 23 rows containing non-finite values (stat_smooth).Warning: Removed 23 rows containing missing values (geom_rect).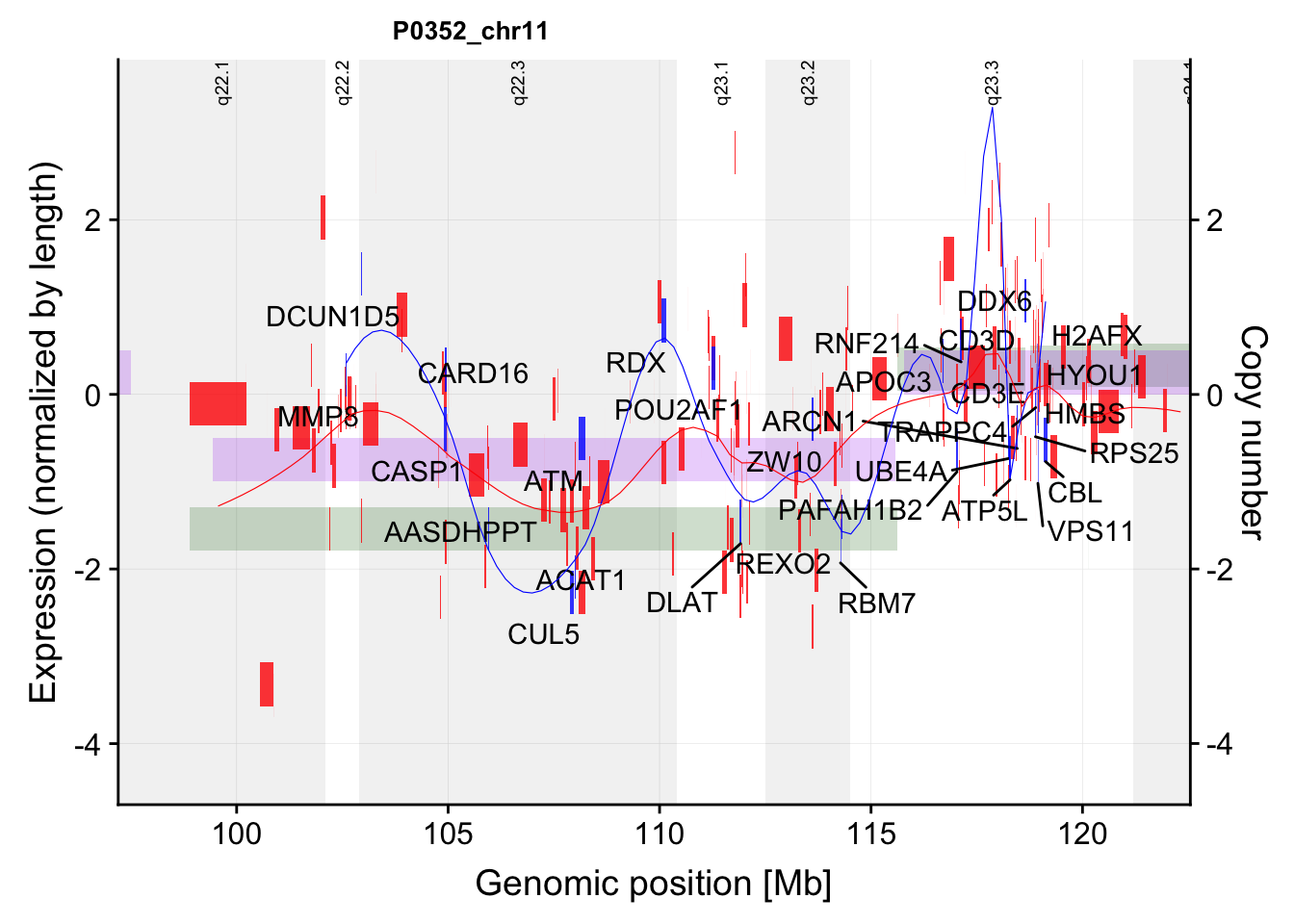
g <- plotExprCNV("P0352","chr11",allBand,allLine, allProtTab, allRnaTab, ifTrend = TRUE,
startPos = 97.2, endPos = 122.55, showLabel = "protein-rna",plotDiff = TRUE)Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelg$main`geom_smooth()` using formula 'y ~ x'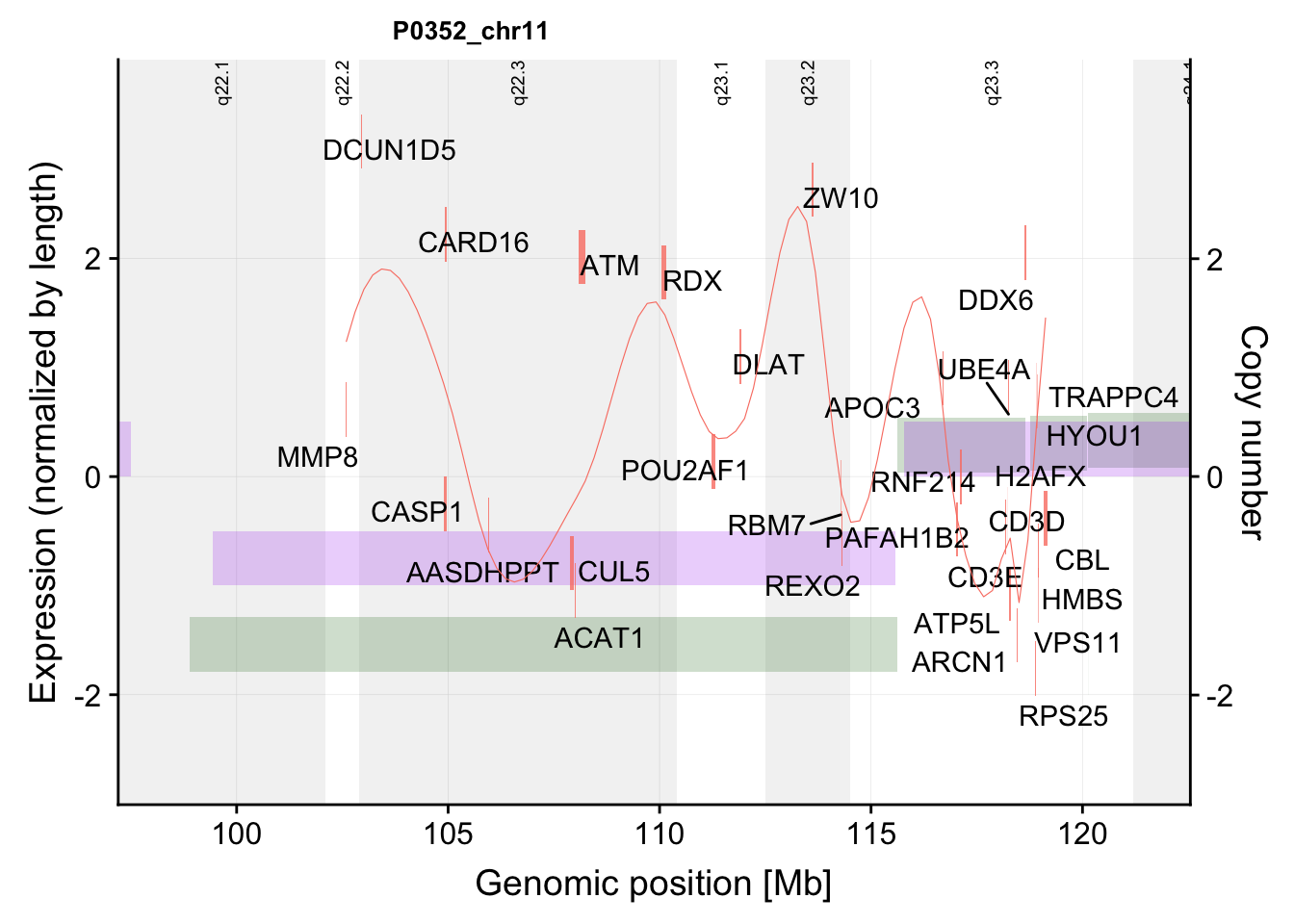
g <- plotExprCNV("P0352","chr11",allBand,allLine, allProtTab, allRnaTab, ifTrend = TRUE,
showLabel = "none",plotDiff = TRUE)Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelg$main`geom_smooth()` using formula 'y ~ x'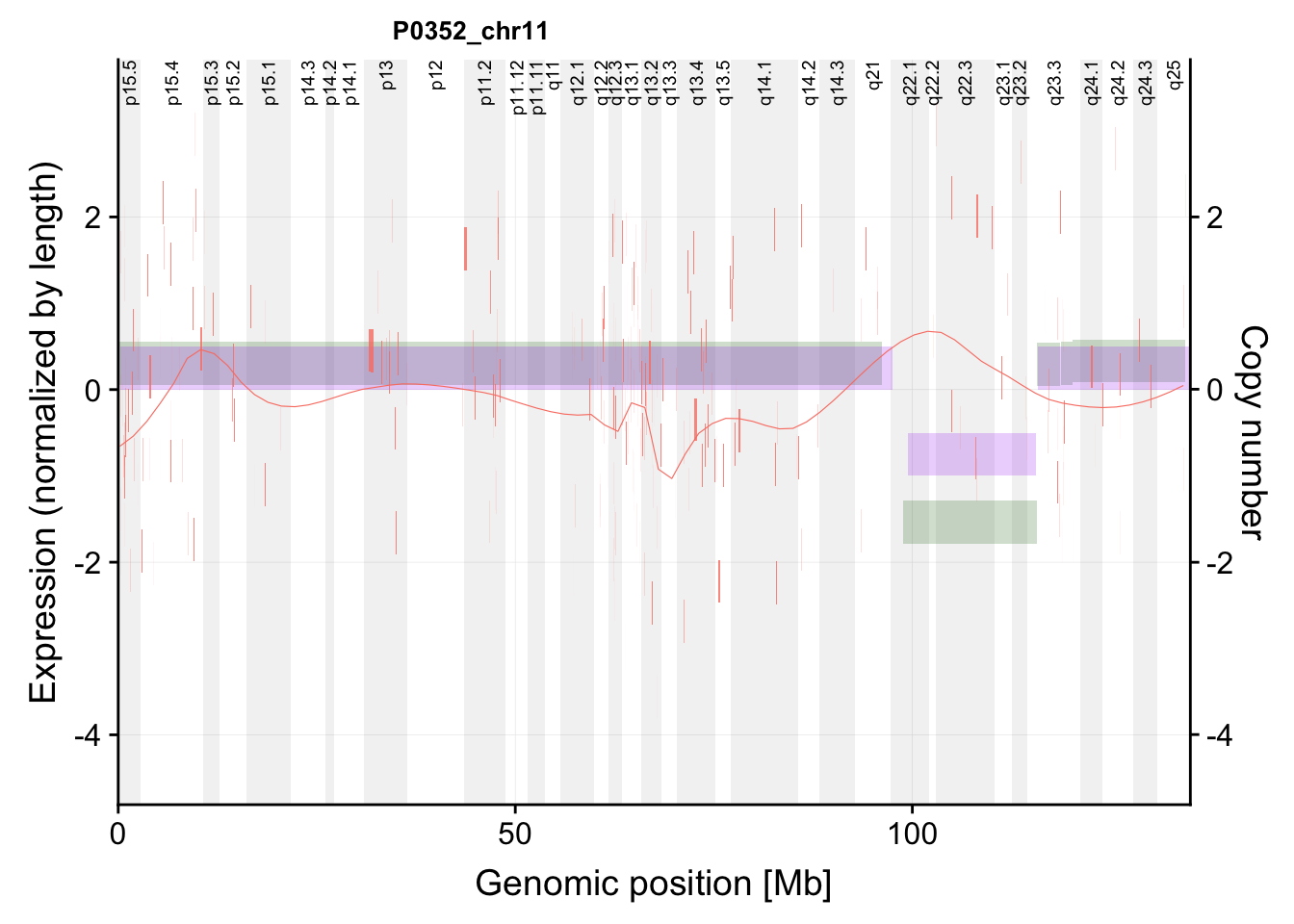
Plot for all patients and all chromosomes (normalized expression)
outDir <- "../public/cnv_plots/"
for (eachPat in unique(allProtTab$patID)) {
pList <- lapply(as.character(unique(allBand$ChromID)), function(eachChr) {
g <- plotExprCNV(eachPat,eachChr,allBand, allLine, allProtTab, allRnaTab, ifTrend = TRUE)
plot_grid(g$main, g$legend, ncol = 1, rel_heights = c(1,0.2))
})
jyluMisc::makepdf(pList, paste0(outDir,eachPat,".pdf"), ncol = 1, nrow = 1,width = 15,height = 6)
}plot for all patients with 11q deletion for the region between q22.1 and q23.3
patList <- intersect(filter(patMeta, del11q %in% 1)$Patient.ID,allProtTab$patID)
pList <- lapply(patList, function(eachPat) {
g <- plotExprCNV(eachPat,"chr11",allBand, allLine, allProtTab, allRnaTab,
ifTrend = TRUE, startPos = 92.8, endPos = 122.55, showLabel = "protein")
plot_grid(g$main, g$legend, ncol = 1, rel_heights = c(1,0.2))
})
jyluMisc::makepdf(pList, "../public/plotCNV_del11q.pdf", ncol = 1, nrow = 1,width = 15,height = 6)Plot for all patients with 11q deletion for the region between q22.1 and q23.3
patList <- intersect(intersect(filter(patMeta, del11q %in% 1)$Patient.ID,allProtTab$patID),allRnaTab$patID)
pList <- lapply(patList, function(eachPat) {
g <- plotExprCNV(eachPat,"chr11",allBand, allLine, allProtTab, allRnaTab,
ifTrend = TRUE, startPos = 92.8, endPos = 122.55, showLabel = "protein-rna",plotDiff = TRUE)
plot_grid(g$main, g$legend, ncol = 1, rel_heights = c(1,0.2))
})
jyluMisc::makepdf(pList, "../public/plotCNV_del11q_diff.pdf", ncol = 1, nrow = 1,width = 15,height = 6)Plot for all patients with 11q deletion for the region between q22.1 and q23.3
patList <- intersect(intersect(filter(patMeta, del11q %in% 1)$Patient.ID,allProtTab$patID),allRnaTab$patID)
pList <- lapply(patList, function(eachPat) {
g <- plotExprCNV(eachPat,"chr11",allBand, allLine, allProtTab, allRnaTab,
ifTrend = TRUE, showLabel = "none",plotDiff = TRUE)
plot_grid(g$main, g$legend, ncol = 1, rel_heights = c(1,0.2))
})
jyluMisc::makepdf(pList, "../public/plotCNV_allChr11_diff.pdf", ncol = 1, nrow = 1,width = 15,height = 6)Plot for summary of all patients with 11q deletion for the region between q22.1 and q23.3
patList <- intersect(intersect(filter(patMeta, del11q %in% 1)$Patient.ID,allProtTab$patID),allRnaTab$patID)
g <- plotExprCNV(patList,"chr11",allBand, allLine, allProtTab, allRnaTab,
ifTrend = TRUE, startPos = 92.8, endPos = 122.55, showLabel = "protein")
pList <- list(plot_grid(g$main, g$legend, ncol = 1, rel_heights = c(1,0.2)))
jyluMisc::makepdf(pList, "../public/plotCNV_del11q_sum.pdf", ncol = 1, nrow = 1,width = 50,height = 10)Compare patients with different genomic background
patBack <- filter(patMeta, Patient.ID %in% unique(allProtTab$patID)) %>%
select(Patient.ID, del17p, trisomy12, del11q, del13p) %>%
rename(patID = Patient.ID) %>%
mutate_all(as.character) %>%
mutate_at(vars(-patID),str_replace, "1","Mut") %>%
mutate_at(vars(-patID),str_replace, "0","WT")plotExprVar <- function(gene, chr, patBack, allBand, allLine, allProtTab, allRnaTab,
region = c(-Inf,Inf),ifTrend = FALSE, normalize = TRUE, maxVal =2, minVal=-2) {
#table for cyto band
bandTab <- filter(allBand, ChromID == chr, chromStart >= region[1], chromEnd <= region[2]) %>%
mutate(chromMid = chromMid)
#table for expression
plotProtTab <- filter(allProtTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
plotRnaTab <- filter(allRnaTab, ChromID == chr, start_position >= region[1], end_position <= region[2]) %>%
mutate_if(is.factor,as.character)
#summarise group mean
plotProtTab <- plotProtTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
plotRnaTab <- plotRnaTab %>%
mutate(group = patBack[match(patID, patBack$patID),][[gene]]) %>%
filter(!is.na(group)) %>%
group_by(id, group) %>% mutate(meanExpr = mean(expr, na.rm=TRUE)) %>%
distinct(group, id,.keep_all = TRUE) %>% ungroup()
xMax <- max(bandTab$chromEnd, na.rm = T)
#main plot for Protein
gPro <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal, ymax=maxVal,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal, hjust =1, angle = 90, size=2.5) +
geom_rect(data = plotProtTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression (normalized by length)") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
Mut = "darkred", WT = "darkgreen")) +
scale_color_manual(values = c(Mut = "darkred",WT = "darkgreen")) +
ggtitle(paste0("Protein expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10, hjust = 0.3),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gPro <- gPro + geom_smooth(data =filter(plotProtTab, expr >0),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.5,
size =0.2, alpha=0.5)
}
#main plot for RNA
gRna <- ggplot() +
geom_rect(data=bandTab, mapping=aes(xmin=chromStart, xmax=chromEnd, ymin=minVal, ymax=maxVal,
fill=Colour, label = band), alpha=0.1) +
geom_text(data=bandTab, mapping=aes(label=band, x=chromMid), y=maxVal, hjust =1, angle = 90, size=2.5) +
geom_rect(data = plotRnaTab,
mapping=aes(xmin=start_position,
xmax=end_position, ymin=meanExpr, ymax=meanExpr+0.1,
fill = group, label = symbol)) +
scale_x_continuous(expand=c(0,0),limits = c(0,xMax)) +
xlab("Genomic position [Mb]") +
ylab("Expression (normalized by length)") +
scale_fill_manual(values = c(even = "white",odd = "grey50",
Mut = "darkred", WT = "darkgreen")) +
scale_color_manual(values = c(Mut = "darkred",WT = "darkgreen")) +
ggtitle(paste0("RNA expression","(",chr,")")) +
theme(plot.title = element_text(face = "bold", size = 10, hjust = 0.3),
legend.position = "none",
panel.background = element_blank(),
panel.grid.major = element_line(colour="grey90", size=0.1))
if (ifTrend) {
gRna <- gRna + geom_smooth(data =filter(plotRnaTab),
mapping = aes(y=meanExpr, x= mid_position,
color = group),
formula = y ~ x, method = "loess", se=FALSE, span=0.2,
size =0.2, alpha=0.5)
}
#for legend
## if the patient has CNV data
lgTab <- tibble(x= seq(6),y=seq(6),
Expression = c(rep("Mut",3), rep("WT",3)))
lg <- ggplot(lgTab, aes(x=x,y=y)) +
geom_point(aes(fill = Expression), shape =22,size=3) +
scale_fill_manual(values = c(Mut = "darkred", WT = "darkgreen"), name = gene) +
theme(legend.position = "bottom")
lg <- get_legend(lg)
return(list(plotPro = gPro, plotRNA = gRna, legend = lg))
}Trisomy12
pdf("../public/trisomy12_norm.pdf",height = 8, width = 10)
g <- plotExprVar("trisomy12","chr12",patBack,allBand, allLine, allProtTab, allRnaTab, ifTrend = TRUE)Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelplot_grid(g$plotRNA, g$plotPro, g$legend, ncol = 1, rel_heights = c(1,1,0.2))Warning: Removed 222 rows containing non-finite values (stat_smooth).Warning: Removed 222 rows containing missing values (geom_rect).dev.off()quartz_off_screen
2 Del11q
pdf("../public/del11q_norm.pdf",height = 8, width = 10)
g <- plotExprVar("del11q","chr11",patBack,allBand, allLine, allProtTab, allRnaTab, ifTrend = TRUE)Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: label
Warning: Ignoring unknown aesthetics: labelplot_grid(g$plotRNA, g$plotPro, g$legend, ncol = 1, rel_heights = c(1,1,0.2))Warning: Removed 472 rows containing non-finite values (stat_smooth).Warning: Removed 472 rows containing missing values (geom_rect).dev.off()quartz_off_screen
2 Does protein and rna expression correlate less in the del11q region?
load("../output/exprCNV.RData")protExprTab <- filter(allProtTab, ChromID == "chr11")
rnaExprTab <- filter(allRnaTab,ChromID == "chr11")
compareTab <- left_join(protExprTab, select(rnaExprTab, id, patID, expr),by=c("id","patID")) %>%
dplyr::rename(exprProt = expr.x, exprRna=expr.y) %>% filter(!is.na(exprProt),!is.na(exprRna))Warning: Column `id` joining factors with different levels, coercing to
character vectorWarning: Column `patID` joining factors with different levels, coercing to
character vectorSelect regions that deleted in most of the samples (11q22.3 and 11q23.1)
filter(allBand,ChromID == "chr11", band %in% c("q22.3","q23.1")) ChromID chromStart chromEnd band gieStain Colour chromMid
1 chr11 102.9 110.4 q22.3 gpos100 odd 106.65
2 chr11 110.4 112.5 q23.1 gneg even 111.45startPos <- 102.9
endPos <- 112.5
compareTab <- mutate(compareTab, ifDel = ifelse(mid_position >= startPos & mid_position <= endPos,TRUE, FALSE))Correlation test for each protein-rna pair on chr11
resTab <- group_by(compareTab, id) %>% nest() %>%
mutate(m = map(data, ~cor.test(~exprProt + exprRna,.))) %>%
mutate(res = map(m, broom::tidy)) %>% unnest(res) %>%
select(-data, -m) %>%
left_join(distinct(compareTab, id, symbol, ifDel),by = "id")Plot distribution of correlations coefficient for prtein-rna pairs inside and outside of deleted retions
ggplot(resTab, aes(x=estimate, fill = ifDel)) + geom_histogram(position = "identity",alpha =0.5)`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.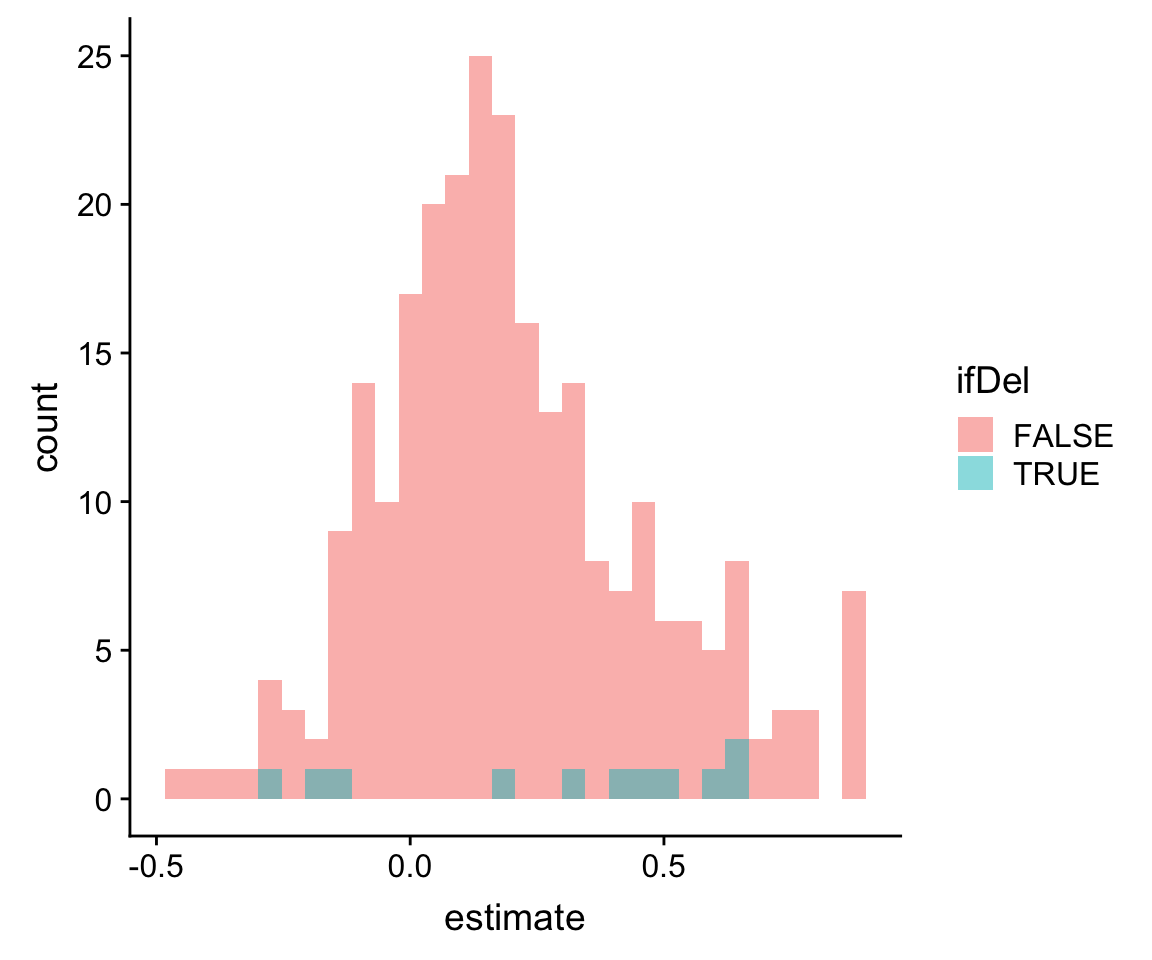
T-test
t.test(estimate~ifDel, resTab)
Welch Two Sample t-test
data: estimate by ifDel
t = -0.84851, df = 10.513, p-value = 0.4151
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-0.3188122 0.1421195
sample estimates:
mean in group FALSE mean in group TRUE
0.2020617 0.2904080 No difference of correlation coefficients
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.4
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] gridExtra_2.3 forcats_0.4.0
[3] stringr_1.4.0 dplyr_0.8.5
[5] purrr_0.3.3 readr_1.3.1
[7] tidyr_1.0.0 tibble_3.0.0
[9] tidyverse_1.3.0 cowplot_0.9.4
[11] ggplot2_3.3.0 DESeq2_1.24.0
[13] SummarizedExperiment_1.14.0 DelayedArray_0.10.0
[15] BiocParallel_1.18.0 matrixStats_0.54.0
[17] Biobase_2.44.0 GenomicRanges_1.36.0
[19] GenomeInfoDb_1.20.0 IRanges_2.18.1
[21] S4Vectors_0.22.0 BiocGenerics_0.30.0
[23] biomaRt_2.40.0
loaded via a namespace (and not attached):
[1] readxl_1.3.1 backports_1.1.4 fastmatch_1.1-0
[4] Hmisc_4.2-0 drc_3.0-1 jyluMisc_0.1.5
[7] workflowr_1.6.0 igraph_1.2.4.1 shinydashboard_0.7.1
[10] splines_3.6.0 TH.data_1.0-10 digest_0.6.19
[13] htmltools_0.4.0 gdata_2.18.0 magrittr_1.5
[16] checkmate_2.0.0 memoise_1.1.0 cluster_2.1.0
[19] openxlsx_4.1.0.1 limma_3.40.2 annotate_1.62.0
[22] modelr_0.1.5 sandwich_2.5-1 piano_2.0.2
[25] prettyunits_1.0.2 colorspace_1.4-1 ggrepel_0.8.1
[28] blob_1.1.1 rvest_0.3.5 haven_2.2.0
[31] xfun_0.8 crayon_1.3.4 RCurl_1.95-4.12
[34] jsonlite_1.6 genefilter_1.66.0 survival_2.44-1.1
[37] zoo_1.8-6 glue_1.3.2 survminer_0.4.4
[40] gtable_0.3.0 zlibbioc_1.30.0 XVector_0.24.0
[43] car_3.0-3 abind_1.4-5 scales_1.1.0
[46] mvtnorm_1.0-11 relations_0.6-8 DBI_1.0.0
[49] Rcpp_1.0.1 plotrix_3.7-6 cmprsk_2.2-8
[52] xtable_1.8-4 progress_1.2.2 htmlTable_1.13.1
[55] foreign_0.8-71 bit_1.1-14 km.ci_0.5-2
[58] Formula_1.2-3 DT_0.7 htmlwidgets_1.3
[61] httr_1.4.1 fgsea_1.10.0 gplots_3.0.1.1
[64] RColorBrewer_1.1-2 acepack_1.4.1 ellipsis_0.2.0
[67] farver_2.0.3 pkgconfig_2.0.2 XML_3.98-1.20
[70] nnet_7.3-12 dbplyr_1.4.2 locfit_1.5-9.1
[73] labeling_0.3 tidyselect_1.0.0 rlang_0.4.5
[76] later_0.8.0 AnnotationDbi_1.46.0 visNetwork_2.0.7
[79] munsell_0.5.0 cellranger_1.1.0 tools_3.6.0
[82] cli_1.1.0 generics_0.0.2 RSQLite_2.1.1
[85] broom_0.5.2 evaluate_0.14 yaml_2.2.0
[88] knitr_1.23 bit64_0.9-7 fs_1.4.0
[91] zip_2.0.2 survMisc_0.5.5 caTools_1.17.1.2
[94] nlme_3.1-140 mime_0.7 slam_0.1-45
[97] xml2_1.2.2 compiler_3.6.0 rstudioapi_0.10
[100] curl_3.3 ggsignif_0.5.0 marray_1.62.0
[103] reprex_0.3.0 geneplotter_1.62.0 stringi_1.4.3
[106] lattice_0.20-38 Matrix_1.2-17 KMsurv_0.1-5
[109] shinyjs_1.0 vctrs_0.2.4 pillar_1.4.3
[112] lifecycle_0.2.0 data.table_1.12.2 bitops_1.0-6
[115] httpuv_1.5.1 R6_2.4.0 latticeExtra_0.6-28
[118] promises_1.0.1 KernSmooth_2.23-15 rio_0.5.16
[121] codetools_0.2-16 MASS_7.3-51.4 gtools_3.8.1
[124] exactRankTests_0.8-30 assertthat_0.2.1 rprojroot_1.3-2
[127] withr_2.1.2 multcomp_1.4-10 GenomeInfoDbData_1.2.1
[130] mgcv_1.8-28 hms_0.5.2 grid_3.6.0
[133] rpart_4.1-15 rmarkdown_1.13 carData_3.0-2
[136] ggpubr_0.2.1 git2r_0.26.1 maxstat_0.7-25
[139] sets_1.0-18 shiny_1.3.2 lubridate_1.7.4
[142] base64enc_0.1-3