Raw data processing of the new proteomic data from timsTOF machine
Junyan Lu
2020-08-06
Last updated: 2020-08-06
Checks: 6 1
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you'll want to first commit it to the Git repo. If you're still working on the analysis, you can ignore this warning. When you're finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it's best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3fb50c5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/analysisDrugResponses_IC50_cache/
Ignored: analysis/analysisDrugResponses_cache/
Ignored: analysis/complexAnalysis_IGHV_alternative_cache/
Ignored: analysis/complexAnalysis_IGHV_cache/
Ignored: analysis/complexAnalysis_trisomy12_alteredPQR_cache/
Ignored: analysis/complexAnalysis_trisomy12_alternative_cache/
Ignored: analysis/complexAnalysis_trisomy12_cache/
Ignored: analysis/correlateCLLPD_cache/
Ignored: analysis/correlateRNAexpression_cache/
Ignored: analysis/manuscript_S1_Overview_cache/
Ignored: analysis/manuscript_S2_genomicAssociation_cache/
Ignored: analysis/manuscript_S3_trisomy12_cache/
Ignored: analysis/manuscript_S4_IGHV_cache/
Ignored: analysis/manuscript_S5_trisomy19_cache/
Ignored: analysis/manuscript_S6_del11q_cache/
Ignored: analysis/manuscript_S7_SF3B1_cache/
Ignored: analysis/manuscript_S8_drugResponse_Outcomes_cache/
Ignored: analysis/predictOutcome_cache/
Ignored: code/.Rhistory
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: analysis/.trisomy12_norm.pdf
Untracked: analysis/CNVanalysis_11q.Rmd
Untracked: analysis/CNVanalysis_trisomy12.Rmd
Untracked: analysis/CNVanalysis_trisomy19.Rmd
Untracked: analysis/SUGP1_splicing.svg.xml
Untracked: analysis/analysisDrugResponses.Rmd
Untracked: analysis/analysisDrugResponses_IC50.Rmd
Untracked: analysis/analysisPCA.Rmd
Untracked: analysis/analysisSplicing.Rmd
Untracked: analysis/analysisTrisomy19.Rmd
Untracked: analysis/annotateCNV.Rmd
Untracked: analysis/complexAnalysis_IGHV.Rmd
Untracked: analysis/complexAnalysis_IGHV_alternative.Rmd
Untracked: analysis/complexAnalysis_overall.Rmd
Untracked: analysis/complexAnalysis_trisomy12.Rmd
Untracked: analysis/complexAnalysis_trisomy12_alternative.Rmd
Untracked: analysis/correlateGenomic_PC12adjusted.Rmd
Untracked: analysis/correlateGenomic_noBlock.Rmd
Untracked: analysis/correlateGenomic_noBlock_MCLL.Rmd
Untracked: analysis/correlateGenomic_noBlock_UCLL.Rmd
Untracked: analysis/correlateRNAexpression.Rmd
Untracked: analysis/default.css
Untracked: analysis/del11q.pdf
Untracked: analysis/del11q_norm.pdf
Untracked: analysis/manuscript_S0_PrepareData.Rmd
Untracked: analysis/manuscript_S1_Overview.Rmd
Untracked: analysis/manuscript_S2_genomicAssociation.Rmd
Untracked: analysis/manuscript_S3_trisomy12.Rmd
Untracked: analysis/manuscript_S4_IGHV.Rmd
Untracked: analysis/manuscript_S5_trisomy19.Rmd
Untracked: analysis/manuscript_S6_del11q.Rmd
Untracked: analysis/manuscript_S7_SF3B1.Rmd
Untracked: analysis/manuscript_S8_drugResponse_Outcomes.Rmd
Untracked: analysis/peptideValidate.Rmd
Untracked: analysis/plotExpressionCNV.Rmd
Untracked: analysis/processPeptides_LUMOS.Rmd
Untracked: analysis/processProteomics_timsTOF_Hela.Rmd
Untracked: analysis/processProteomics_timsTOF_new.Rmd
Untracked: analysis/style.css
Untracked: analysis/test.pdf
Untracked: analysis/test.svg
Untracked: analysis/trisomy12.pdf
Untracked: analysis/trisomy12_AFcor.Rmd
Untracked: analysis/trisomy12_norm.pdf
Untracked: code/AlteredPQR.R
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/200725_cll_diaPASEF_direct_reports/
Untracked: data/200728_cll_diaPASEF_direct_plus_hela_reports/
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/LUMOS_protAnnotation_fix.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/example_proteomics_data
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/proteins_in_complexes
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/Fig1A.pdf
Untracked: output/Fig1A.png
Untracked: output/Fig1A.pptx
Untracked: output/LUMOS_processed.RData
Untracked: output/MSH6_splicing.svg
Untracked: output/SUGP1_splicing.eps
Untracked: output/SUGP1_splicing.pdf
Untracked: output/SUGP1_splicing.svg
Untracked: output/cnv_plots.zip
Untracked: output/cnv_plots/
Untracked: output/cnv_plots_norm.zip
Untracked: output/ddsrna_enc.RData
Untracked: output/deResList.RData
Untracked: output/deResList_timsTOF.RData
Untracked: output/dxdCLL.RData
Untracked: output/dxdCLL2.RData
Untracked: output/encMap.RData
Untracked: output/exprCNV.RData
Untracked: output/exprCNV_enc.RData
Untracked: output/lassoResults_CPS.RData
Untracked: output/lassoResults_IC50.RData
Untracked: output/patMeta_enc.RData
Untracked: output/pepCLL_lumos.RData
Untracked: output/pepCLL_lumos_enc.RData
Untracked: output/pepTab_lumos.RData
Untracked: output/pheno1000_enc.RData
Untracked: output/pheno1000_main.RData
Untracked: output/plotCNV_allChr11_diff.pdf
Untracked: output/plotCNV_del11q_sum.pdf
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_LUMOS_20200320.RData
Untracked: output/proteomic_LUMOS_20200430.RData
Untracked: output/proteomic_LUMOS_enc.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/proteomic_timsTOF_enc.RData
Untracked: output/proteomic_timsTOF_new_20200806.RData
Untracked: output/splicingResults.RData
Untracked: output/survival_enc.RData
Untracked: output/timsTOF_processed.RData
Untracked: plotCNV_del11q_diff.pdf
Untracked: supp_latex/
Unstaged changes:
Modified: analysis/_site.yml
Modified: analysis/analysisSF3B1.Rmd
Modified: analysis/compareProteomicsRNAseq.Rmd
Modified: analysis/correlateCLLPD.Rmd
Modified: analysis/correlateGenomic.Rmd
Deleted: analysis/correlateGenomic_removePC.Rmd
Modified: analysis/correlateMIR.Rmd
Modified: analysis/correlateMethylationCluster.Rmd
Modified: analysis/index.Rmd
Modified: analysis/predictOutcome.Rmd
Modified: analysis/processProteomics_LUMOS.Rmd
Modified: analysis/qualityControl_LUMOS.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Data input
Read and annotate data
Read in data and annotate raw data (unnormalized)
rawTab <- read_tsv("../data/200725_cll_diaPASEF_direct_reports/20200727_104510_200725_cll_diaPASEF_direct_Report_Protein_long.tsv") %>%
select(R.Replicate, PG.ProteinAccessions, PG.ProteinNames,PG.Quantity) %>%
dplyr::rename(id = R.Replicate, name = PG.ProteinAccessions,
count = PG.Quantity, protName = PG.ProteinNames) %>%
mutate(id = paste0("A_1_",id),
protName = str_replace_all(protName,"_HUMAN","")) %>%
filter(!is.na(count))
#annotate patient ID
patAnno <- readxl::read_xlsx("../data/SampleAnnotation_cleaned.xlsx") %>%
mutate(id = paste0("A_1_",id)) %>%
select(-Institute, -Source, -diagnosis)
#annotate basic genomic feature
genAnno <- patMeta %>% select(Patient.ID, gender, IGHV.status, trisomy12)
#annotate technical variable
techTab <- readxl::read_xlsx("../data/20191025_Proteom_submitted_samples_final.xlsx") %>%
select(`Patient ID`, operator, viability, batch, `date of sample processing`, `protein conc. in ug`, `freeze-thaw cycles of peptide solution`) %>% dplyr::rename(patID = `Patient ID`, processDate = `date of sample processing`, proteinConc = `protein conc. in ug`, `freeThawCycle` = `freeze-thaw cycles of peptide solution`) %>%
mutate(batch = ifelse(batch == "test run", "0", batch))
patAnno <- left_join(patAnno, genAnno, by = c(patID = "Patient.ID")) %>%
left_join(techTab, by = "patID")
rawTab <- left_join(rawTab, patAnno, by = "id")Generate formated protein ID
idMap <- tibble(name = unique(rawTab$name)) %>%
mutate(ID = paste0("prot",seq(nrow(.))))
rawTab <- left_join(rawTab, idMap, by = "name")Create SummarizedExperiment object
protCLL <- tidyToSum(rawTab, "ID", "patID", "count",
annoCol = colnames(patAnno)[colnames(patAnno) != "patID"],
annoRow = c("name","protName"))
rowData(protCLL)$ID <- rownames(protCLL)Dimension of the original data
dim(protCLL)[1] 5196 50Initial QA
Check distribution of missing values
Pattern of missing values
plot_missval(protCLL) Some samples show less detection rate than others.
Some samples show less detection rate than others.
Frequencies of missing values for each sample
proTab <- sumToTiday(protCLL, "uniprotID", "patientID")
patMiss <- group_by(proTab, patientID) %>%
summarise(freqNA = sum(is.na(count))/length(count)) %>%
arrange(desc(freqNA)) %>%
mutate(patientID = factor(patientID, levels = patientID))`summarise()` ungrouping output (override with `.groups` argument)ggplot(patMiss, aes(x = patientID, y = freqNA)) + geom_point(size=3) +
geom_segment(aes(x=patientID, xend=patientID, y=0, yend=freqNA)) +
theme(axis.text.x = element_text(angle = 90, vjust =0.5, hjust=1)) + ylab("Frenquency")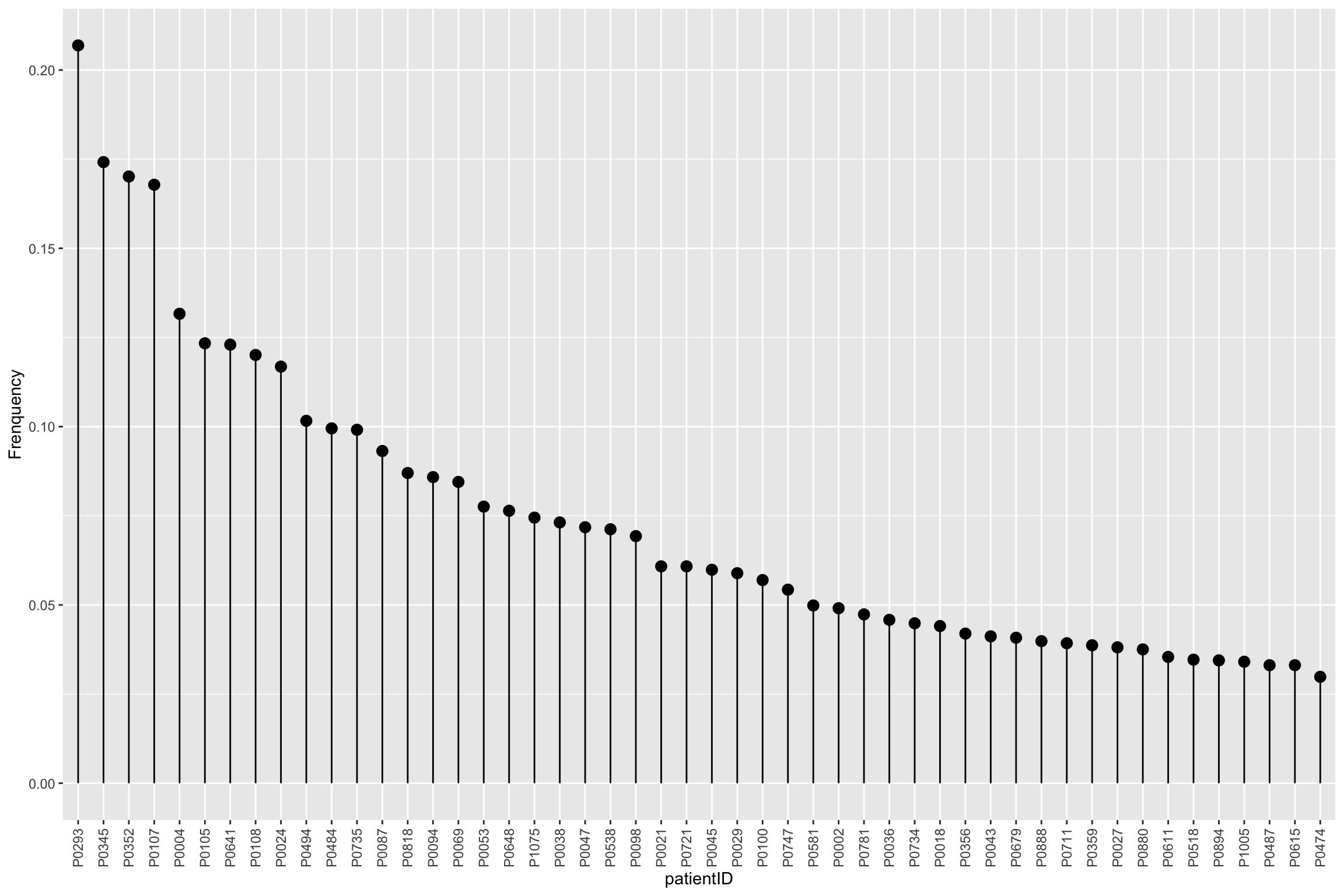
Proteins with missing values
proMiss <- group_by(proTab, uniprotID) %>%
summarise(freqNA = sum(is.na(count))/length(count)) %>%
arrange(desc(freqNA)) %>%
mutate(uniprotID = factor(uniprotID, levels = uniprotID))`summarise()` ungrouping output (override with `.groups` argument)head(proMiss)# A tibble: 6 x 2
uniprotID freqNA
<fct> <dbl>
1 prot5179 0.98
2 prot5183 0.98
3 prot5187 0.98
4 prot5191 0.98
5 prot5192 0.98
6 prot5195 0.98ggplot(proMiss, aes(x=freqNA)) + geom_histogram() +
xlab("Missing value frequency")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.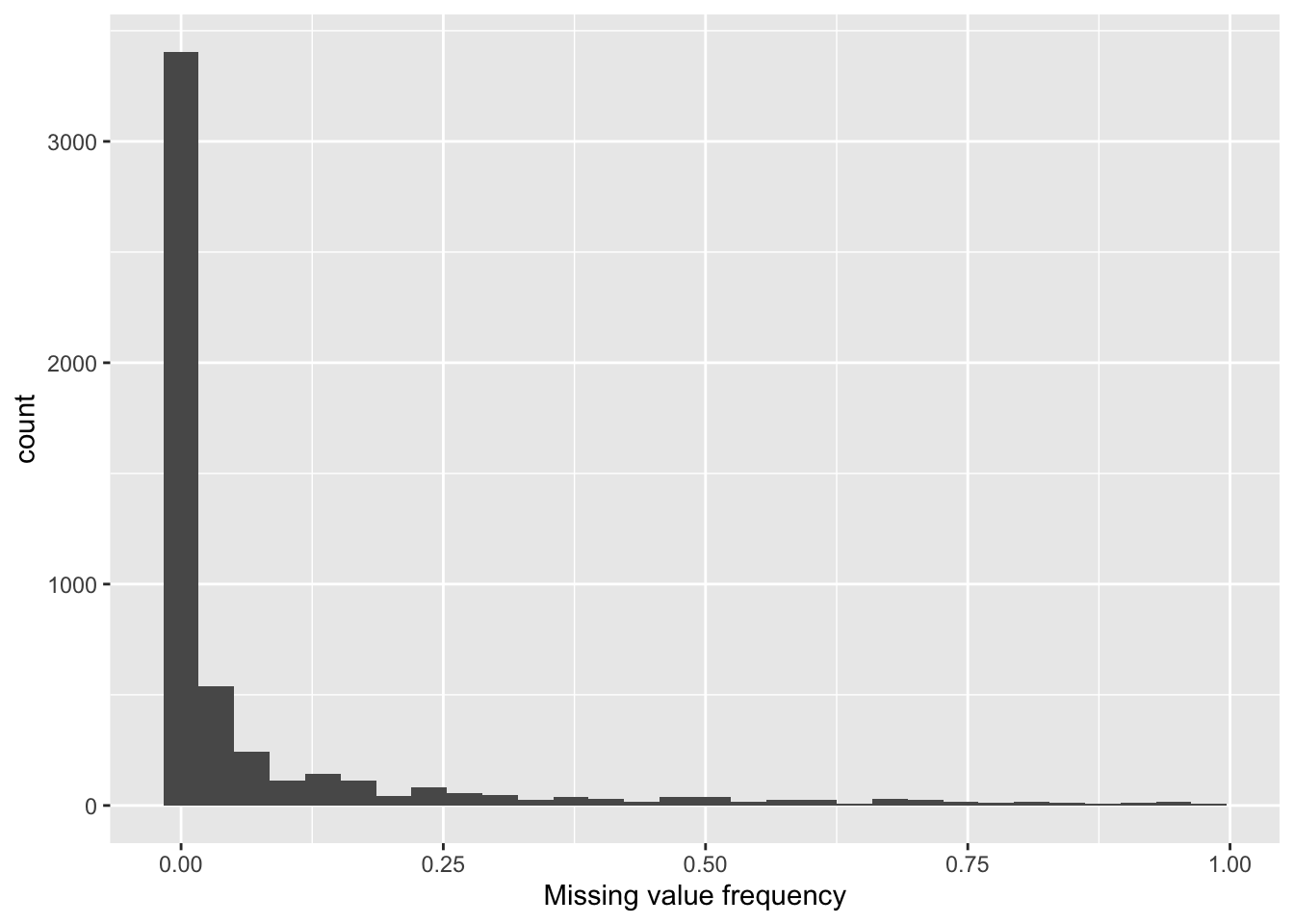
Missing value cut-off versus number of remaining proteins
sumTab <- lapply(seq(0,1,by = 0.01), function(x) tibble(cut = x, freq = sum(proMiss$freqNA < x)/nrow(proMiss))) %>% bind_rows()
ggplot(sumTab, aes(x=cut, y=freq)) + geom_line() + xlab("Missing value cut-off") + ylab("Percent remaining")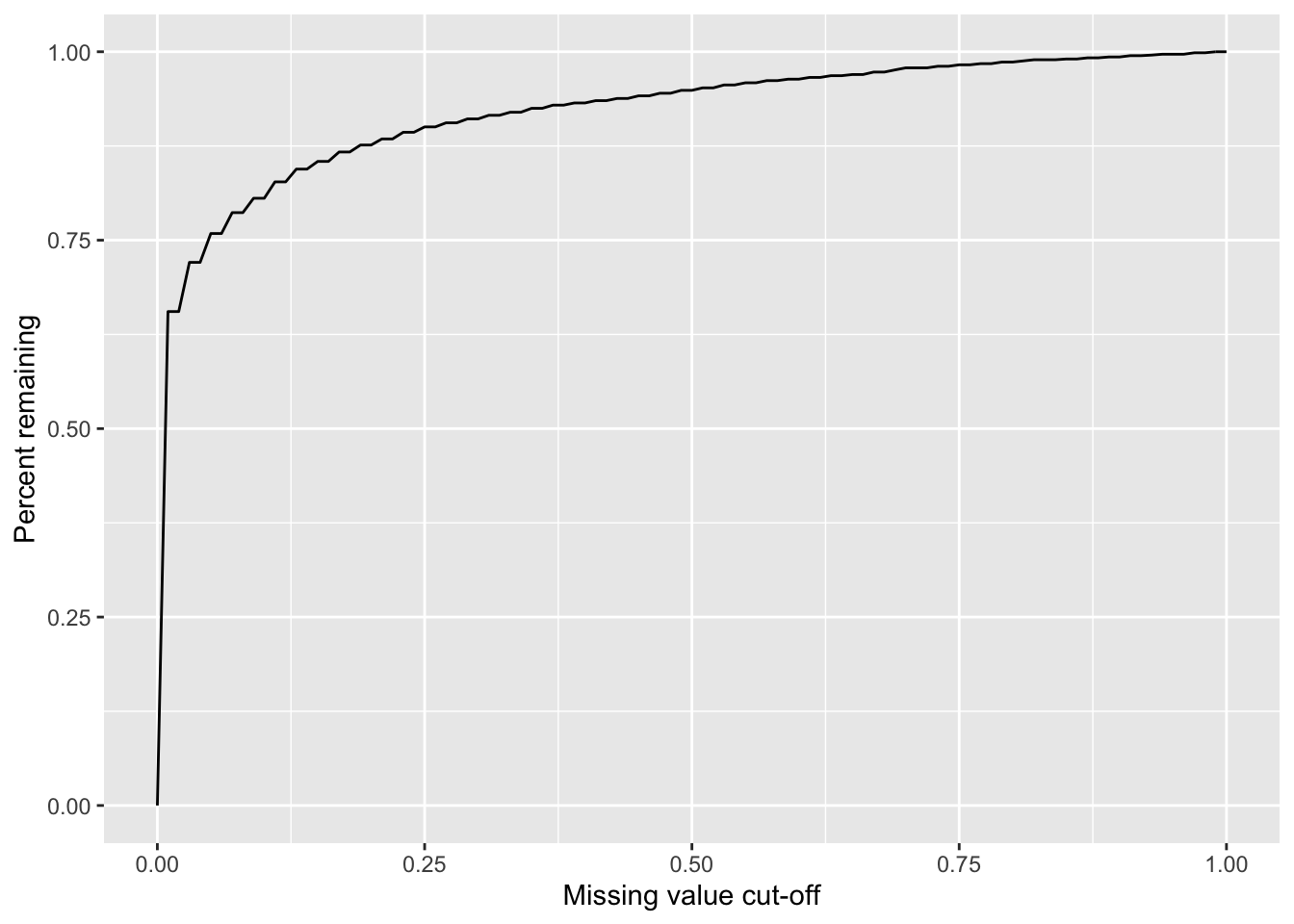
Missing value frequency versus median expression
compareTab <- group_by(proTab, uniprotID) %>%
summarise(freqNA = sum(is.na(count))/length(count),
medianExpr = median(log2(count), na.rm=TRUE))
ggplot(compareTab, aes(x=freqNA, y = medianExpr)) + geom_point() + geom_smooth(method = "loess") +
ylab("Median log2 count") + xlab("Frequency of missing values")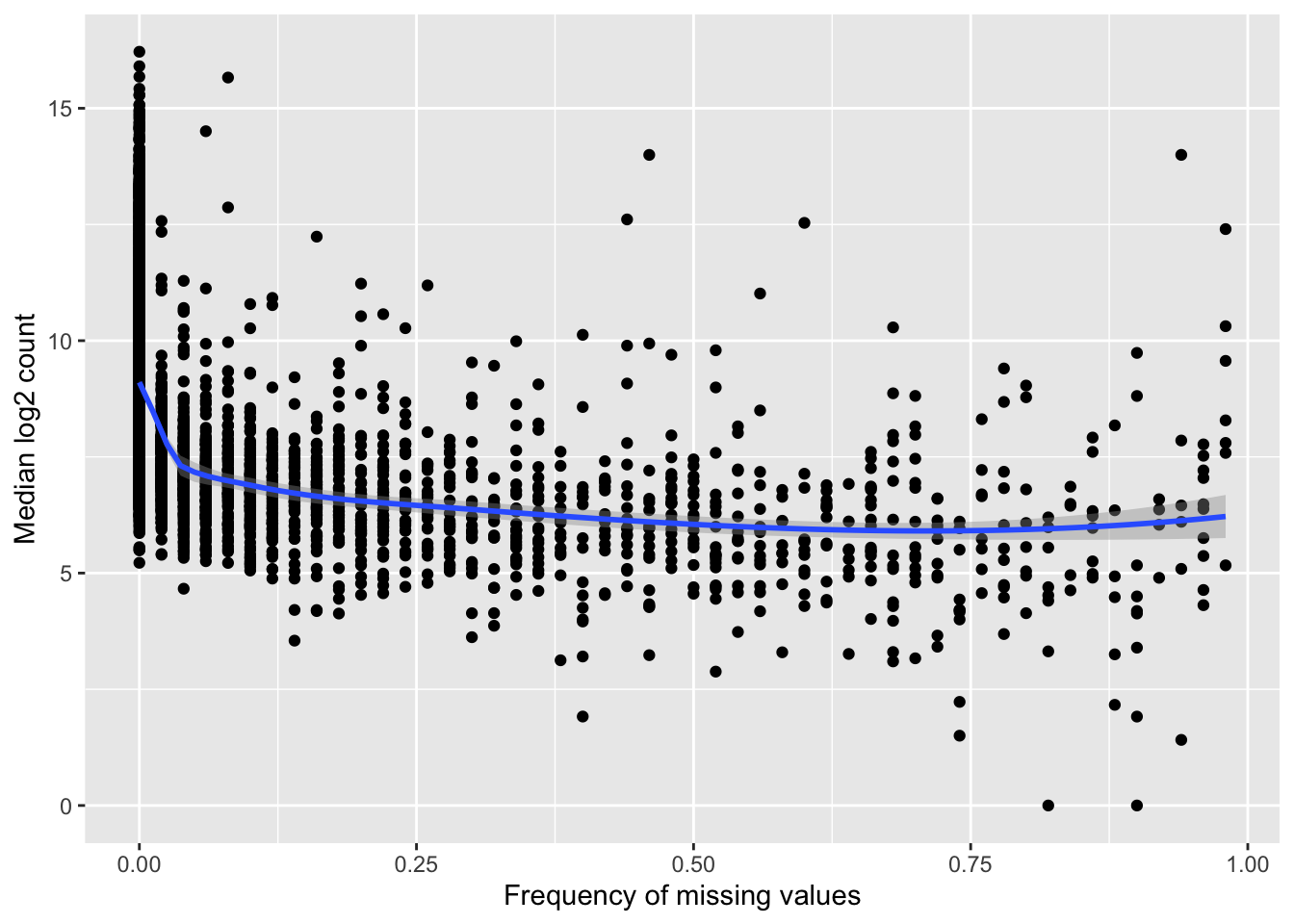 Highly expressed proteins tend to have higher detection rate.
Highly expressed proteins tend to have higher detection rate.
Remove proteins with more than 50% missing values
cut=0.5
protCLL_filt <- protCLL[rowSums(is.na(assay(protCLL)))/ncol(protCLL) <= cut,]Dimension of the filtered data
dim(protCLL_filt)[1] 4947 50Data normalization
Distribution of raw data
protTab <- sumToTiday(protCLL_filt, "id","patientID")
ggplot(protTab, aes(x=patientID, y=count)) + geom_boxplot() + scale_y_log10() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1))Warning: Removed 10294 rows containing non-finite values (stat_boxplot).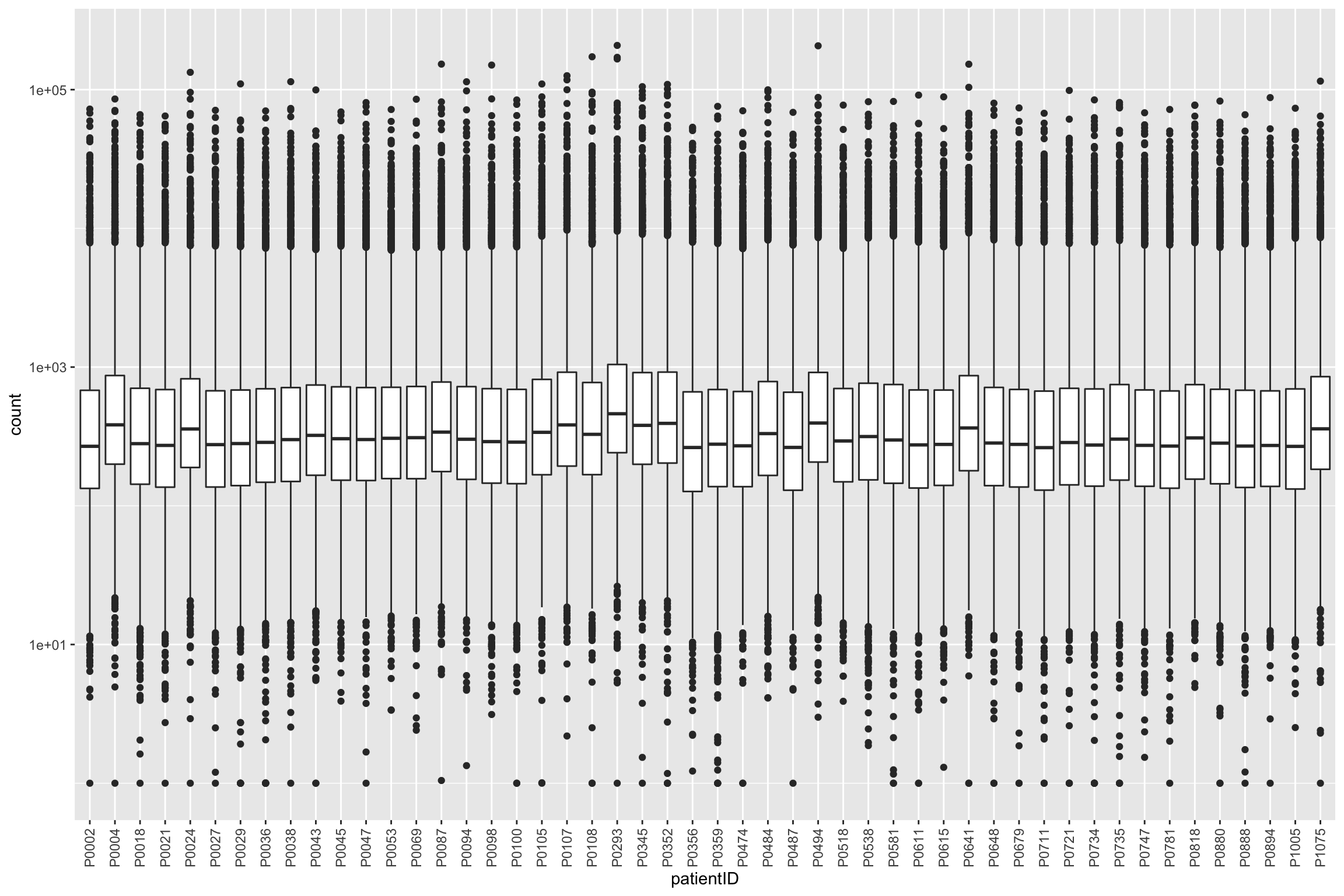
Normalize data using vsn package
exprMat <- assay(protCLL_filt)
resVsn <- vsnMatrix(exprMat)
protCLL_norm <- protCLL_filt
assay(protCLL_norm) <- predict(resVsn, exprMat)Boxplot of normalized counts
protTab <- sumToTiday(protCLL_norm, "uniprotID","patientID")
ggplot(protTab, aes(x=patientID, y=count)) + geom_boxplot() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust=1))Warning: Removed 10294 rows containing non-finite values (stat_boxplot).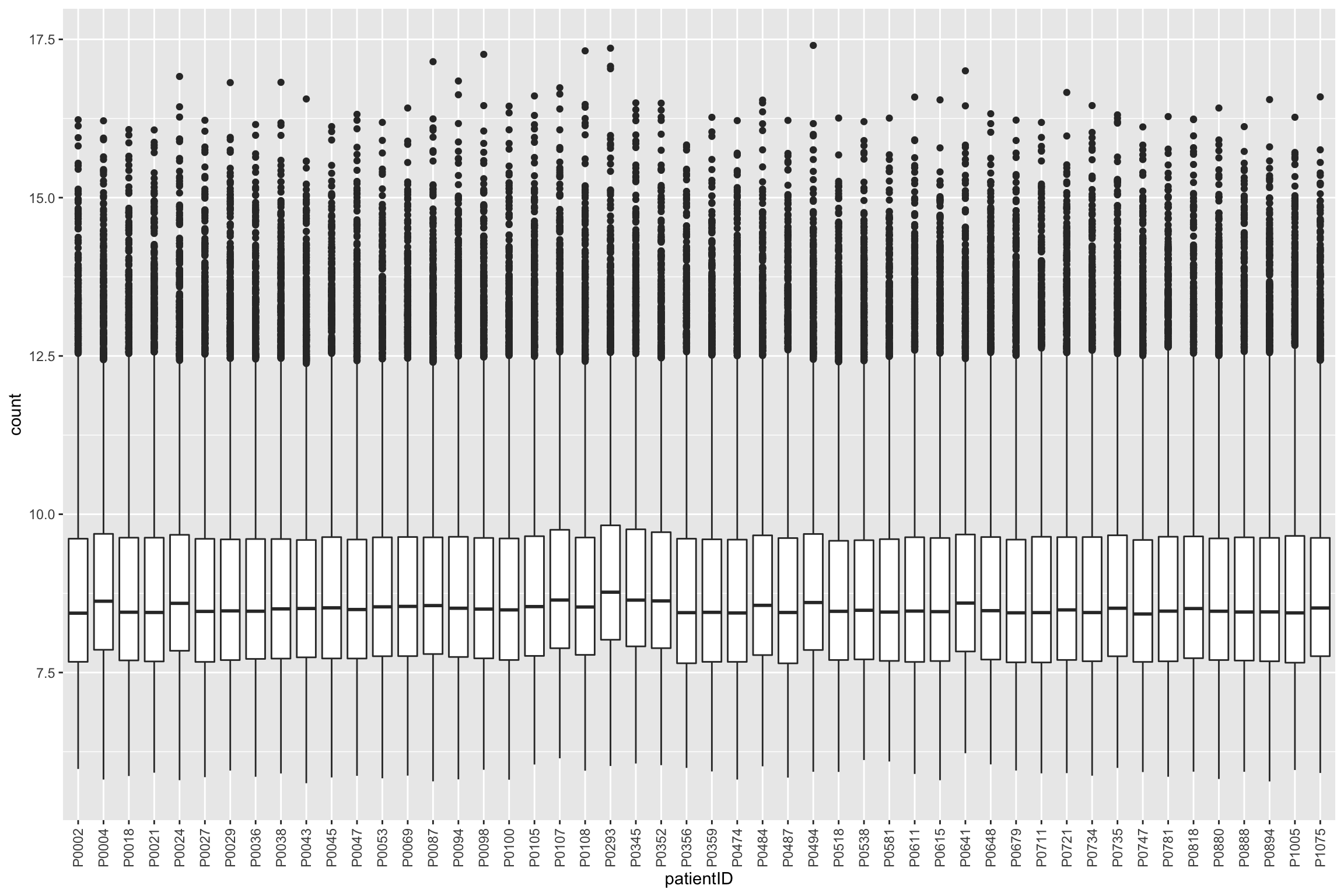
Mean VS SD plot after normalization
vsn::meanSdPlot(resVsn)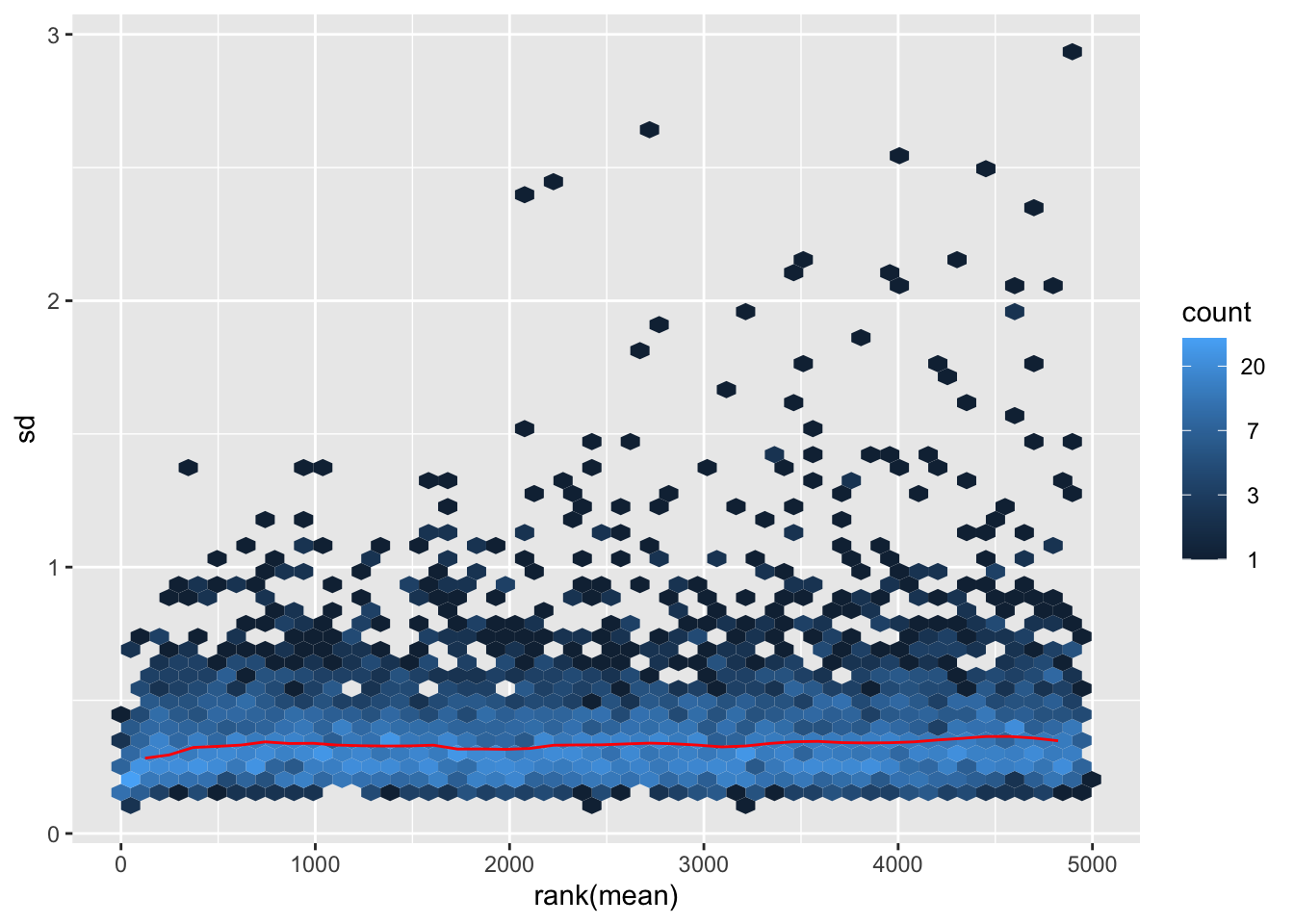 Looks OK. Although lowly expressed proteins still have higher variance.
Looks OK. Although lowly expressed proteins still have higher variance.
Post processing
Impute missing values
For impute missing values, first I need to see whether the data is missing at random or not.
Check missing value pattern again (random or not?)
Missing value pattern after normalization and filtering
plot_missval(protCLL_norm)
Detection rate of proteins with and without missing values
plot_detect(protCLL_norm)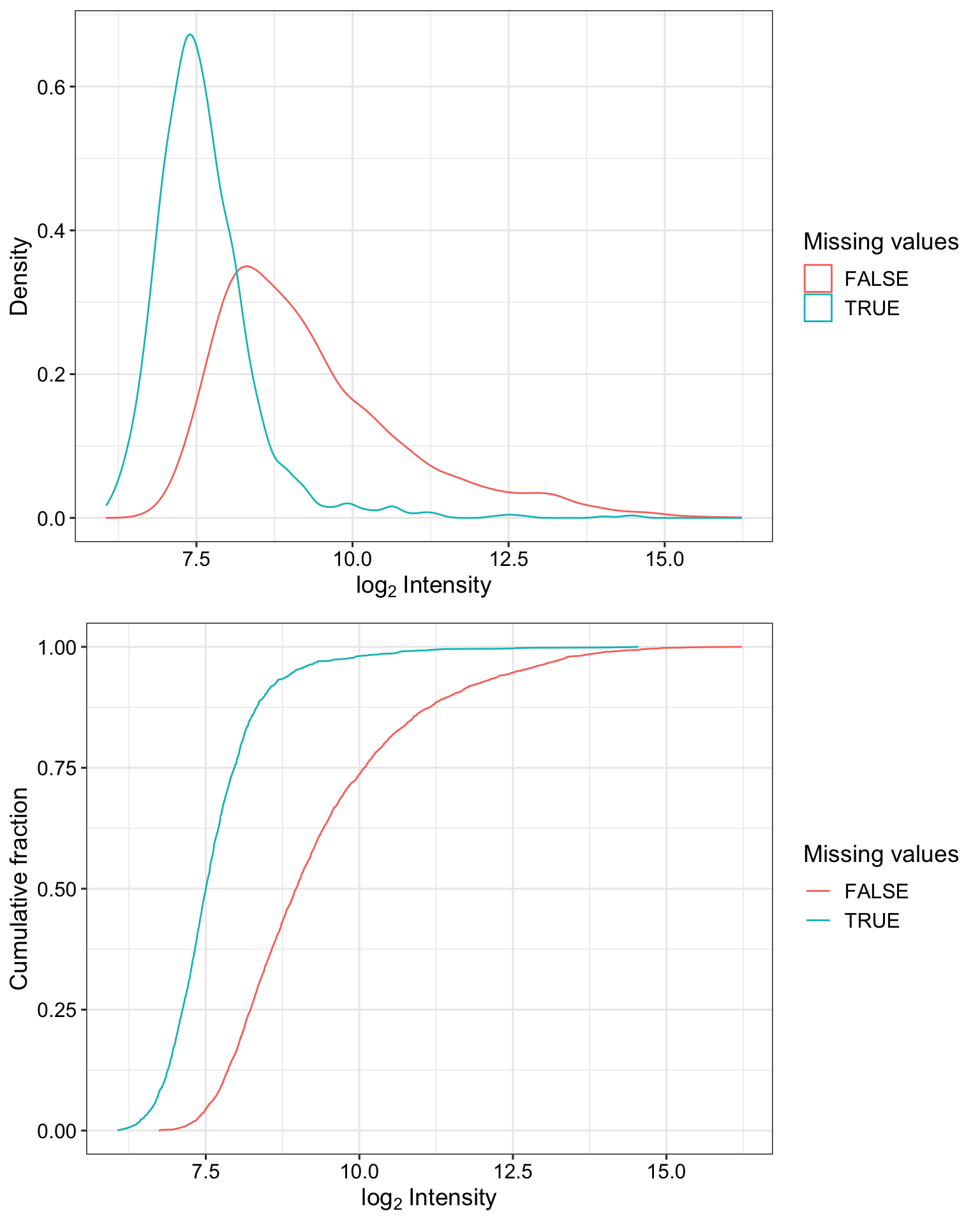 Proteins with missing values have on average low intensities. Not missing at random.
Proteins with missing values have on average low intensities. Not missing at random.
Impute missing values
Impute missing values using quantile regression-based left-censored function (QRILC)
This is a method for imputing missing not at random data.
protCLL_imp <- impute(protCLL_norm, fun = "QRILC")#add QRILC imputed data
assays(protCLL_norm)[["QRILC"]] <- assay(protCLL_imp)Annotate protein information using bioMart
Prepare protein id table
idTab <- map_df(unique(rowData(protCLL)$name), ~tibble(ID = ., uniprotID = str_split(.,";")[[1]]))ensembl = useMart("ensembl",dataset="hsapiens_gene_ensembl",host="grch37.ensembl.org")
ids <- idTab$uniprotID
anno <- getBM(attributes=c('hgnc_symbol','ensembl_gene_id',
'uniprotswissprot'),
filters = 'uniprotswissprot',
values = ids,
mart = ensembl)Warning: `select_()` is deprecated as of dplyr 0.7.0.
Please use `select()` instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_warnings()` to see where this warning was generated.Warning: `filter_()` is deprecated as of dplyr 0.7.0.
Please use `filter()` instead.
See vignette('programming') for more help
This warning is displayed once every 8 hours.
Call `lifecycle::last_warnings()` to see where this warning was generated.Cache foundidTab <- left_join(idTab, anno, by = c(uniprotID="uniprotswissprot")) %>%
mutate(protName = rowData(protCLL)[match(ID, rowData(protCLL)$name),]$protName)Proteins that can not be mapped
filter(idTab, is.na(ensembl_gene_id)) %>% DT::datatable()Map those proteins using uniprotID to symbol list
idTab.miss <- filter(idTab,is.na(ensembl_gene_id))
mapSymbol <- read_tsv("../data/mapSymbol.txt")Parsed with column specification:
cols(
From = col_character(),
To = col_character()
)idTab.miss <- mutate(idTab.miss, hgnc_symbol = mapSymbol[match(uniprotID, mapSymbol$From),]$To)
#proteins that can not be mapped
unique(filter(idTab.miss, is.na(hgnc_symbol))$uniprotID) [1] "A6NFI3" "Q58FF7" "Q6DN03" "Q6DRA6" "Q6SPF0"
[6] "Q7Z739" "O60361" "P57053" "P0DOX8" "Q9BRL6"
[11] "Q9H6W3" "A0A075B6S6" "P06310" "C9J7I0" "Q6ZSR9"
[16] "A0A0B4J2H0" "P01768" "P0DP03" "P0DOX2" "Q9BZK3"
[21] "Q9NQA3" "P0DSE2" "A0A0B4J1U7" "A0A0J9YX35" "P01717"
[26] "P01893" "Q9BZD3" "P0DOX5" "P0DOX7" "P0DP91"
[31] "P0DPB6" #they can be safely removed
idTab.miss <- filter(idTab.miss, !is.na(hgnc_symbol)) %>%
dplyr::select(-ensembl_gene_id)Get ensemble IDs for those proteins using symbol
ids <- idTab.miss$hgnc_symbol
anno <- getBM(attributes=c('hgnc_symbol','ensembl_gene_id'),
filters = 'hgnc_symbol',
values = ids,
mart = ensembl)Cache foundidTab.miss <- left_join(idTab.miss, anno, by = "hgnc_symbol")Genes whose ensembl ID still can not found, use manual annotation
mapSE <- structure(c("ENSG00000180389","ENSG00000282100","ENSG00000180448",
NA,NA,NA,"ENSG00000177144","ENSG00000223614","ENSG00000160221",
"ENSG00000280071","ENSG00000278615","ENSG00000282651",
"ENSG00000275895"),
names=c("ATP5F1EP2","HSP90AB4P","ARHGAP45","IGHV3-43D","GAGE7","IGHV3-30-3","NUDT4B",
"ZNF735","GATD3A","GATD3B","C11orf98","IGHV5-10-1","U2AF1L5"))
idTab.miss <- mutate(idTab.miss, ensembl_gene_id = ifelse(is.na(ensembl_gene_id), mapSE[hgnc_symbol], ensembl_gene_id))Update annotations for proteins with missing annotation
idTab <- bind_rows(filter(idTab, !is.na(ensembl_gene_id)), idTab.miss)Retrieve chromosome information (using ensembl id)
#firstly based on id
ids <- idTab$ensembl_gene_id
anno <- getBM(attributes=c('ensembl_gene_id','chromosome_name'),
filters = 'ensembl_gene_id',
values = ids,
mart = ensembl)Cache foundidTab <- mutate(idTab,
chromosome_name = anno[match(idTab$ensembl_gene_id,anno$ensembl_gene_id),]$chromosome_name) %>%
filter(!grepl("CHR|HG",chromosome_name)) %>%
arrange(ensembl_gene_id) %>%
distinct(uniprotID, .keep_all = TRUE) #some proteins can be mapped to several genes, this is normal and mostly happens to histones. This step will remove then and only keep one. This should be fine.
filter(idTab, is.na(chromosome_name))# A tibble: 5 x 6
ID uniprotID hgnc_symbol ensembl_gene_id protName chromosome_name
<chr> <chr> <chr> <chr> <chr> <chr>
1 P0DN76;Q01081 P0DN76 U2AF1L5 ENSG00000275895 U2AF5;U2… <NA>
2 E9PRG8 E9PRG8 C11orf98 ENSG00000278615 CK098 <NA>
3 A0A0B4J2D5;P… A0A0B4J2D5 GATD3B ENSG00000280071 GAL3B;GA… <NA>
4 Q58FF6 Q58FF6 HSP90AB4P ENSG00000282100 H90B4 <NA>
5 P0DP02 P0DP02 IGHV3-30-3 <NA> HVC33 <NA> For proteins chromosome can not be annotated, manually add chromosome info
chrMap <- structure(c("15","21","11","14","14","21"),
names = c("Q58FF6","A0A0B4J2D5",
"E9PRG8","A0A0J9YXX1",
"P0DP04","P0DN76"))
idTab <- mutate(idTab, chromosome_name = ifelse(is.na(chromosome_name),
chrMap[uniprotID],chromosome_name))Annotate
rawTab <- sumToTiday(protCLL, rowID = "rowID", colID = "patID")
rawTab <- left_join(rawTab, idTab, by = c(name = "ID")) %>%
filter(!is.na(uniprotID)) %>% select(-rowID, -ID)
normTab <- sumToTiday(protCLL_norm, rowID = "rowID", colID = "patID")
normTab <- left_join(normTab, idTab, by = c(name = "ID")) %>%
filter(!is.na(uniprotID)) %>% select(-rowID, -ID)Combine and save objects
Combine table
protCLL_raw <- tidyToSum(rawTab, "uniprotID","patID", values = "count",
annoCol = colnames(patAnno)[colnames(patAnno)!="patID"],
annoRow = c("name","ensembl_gene_id","hgnc_symbol","chromosome_name"))
protCLL <- tidyToSum(normTab, "uniprotID","patID", values = c("count", "QRILC"),
annoCol = colnames(patAnno)[colnames(patAnno)!="patID"],
annoRow = c("name","ensembl_gene_id","hgnc_symbol","chromosome_name"))Annotate proteins that can not be uniquely mapped
#for all proteins
dupTab <- rowData(protCLL_raw) %>% data.frame(stringsAsFactors = FALSE) %>% rownames_to_column("id") %>%
group_by(name) %>% summarise(n=length(id)) %>% filter(n>1)`summarise()` ungrouping output (override with `.groups` argument)rowData(protCLL)$uniqueMap <- ! rowData(protCLL)$name %in% dupTab$name
rowData(protCLL_raw)$uniqueMap <- ! rowData(protCLL_raw)$name %in% dupTab$name
#a list of proteins that can not be uniquely mapped
unique(rowData(protCLL[!rowData(protCLL)$uniqueMap,])$hgnc_symbol) [1] "GATD3B" "SNRPGP15" "WASH3P" "NACA" "TIMM23"
[6] "MYL12B" "RBFOX2" "HIST1H2BK" "KRT6A" "HIST1H2AB"
[11] "PRSS1" "ZFY" "H2AFZ" "HIST1H2AK" "TRAPPC2"
[16] "TRAPPC2P1" "SULT1A3" "SULT1A4" "HSPA1A" "HSPA1B"
[21] "U2AF1L5" "IGLC2" "IGLC3" "CALM1" "CALM2"
[26] "CALM3" "GATD3A" "SNRPB" "ZFX" "MYL12A"
[31] "HIST1H2AD" "KRT6C" "UBE2D1" "ATP5E" "HIST1H2BD"
[36] "DEFA1" "DEFA3" "ARF3" "MAGOH" "SNRPG"
[41] "HIST1H2BI" "RPL39" "GNAS" "SNRPN" "HIST1H3B"
[46] "NOMO3" "ARF1" "H3F3B" "CCZ1B" "CCZ1"
[51] "U2AF1" "BNIP2" "NAB1" "GTF2H2" "NAB2"
[56] "HIST2H2AC" "LGALS9B" "ORMDL2" "RPL39P5" "ALG10"
[61] "ALG10B" "NOMO2" "TIMM23B" "ATP5EP2" "LGALS9C"
[66] "HIST2H2AA3" "MZT2B" "GTF2H2C" "MZT2A" "WASH2P"
[71] "HIST2H3D" "H2AFV" "HIST3H2A" "ATCAY" "PRSS3P2"
[76] "HIST1H2AC" "UBE2E3" "MAGOHB" "HIST1H2AH" "UBE2E2"
[81] "CLIC6" "HIST1H2BN" "HIST1H2AJ" "HIST1H2BM" "HIST1H2BL"
[86] "H2AFJ" "RBFOX1" "CLIC5" "ORMDL1" "UBE2D4" Save object
#for other projects
save(protCLL, protCLL_raw, file = "../../var/proteomic_timsTOF_new_20200806.RData")
#for this project
save(protCLL, protCLL_raw, file = "../output/proteomic_timsTOF_new_20200806.RData")
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.15.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] forcats_0.5.0 stringr_1.4.0
[3] dplyr_1.0.0 purrr_0.3.4
[5] readr_1.3.1 tidyr_1.1.0
[7] tibble_3.0.3 ggplot2_3.3.2
[9] tidyverse_1.3.0 SummarizedExperiment_1.16.1
[11] DelayedArray_0.12.3 BiocParallel_1.20.1
[13] matrixStats_0.56.0 GenomicRanges_1.38.0
[15] GenomeInfoDb_1.22.1 IRanges_2.20.2
[17] S4Vectors_0.24.4 biomaRt_2.42.1
[19] DEP_1.8.0 jyluMisc_0.1.5
[21] vsn_3.54.0 Biobase_2.46.0
[23] BiocGenerics_0.32.0 pheatmap_1.0.12
[25] cowplot_1.0.0 limma_3.42.2
loaded via a namespace (and not attached):
[1] utf8_1.1.4 shinydashboard_0.7.1 gmm_1.6-5
[4] tidyselect_1.1.0 RSQLite_2.2.0 AnnotationDbi_1.48.0
[7] htmlwidgets_1.5.1 grid_3.6.0 norm_1.0-9.5
[10] maxstat_0.7-25 munsell_0.5.0 codetools_0.2-16
[13] preprocessCore_1.48.0 DT_0.14 withr_2.2.0
[16] colorspace_1.4-1 knitr_1.29 rstudioapi_0.11
[19] ggsignif_0.6.0 mzID_1.24.0 labeling_0.3
[22] git2r_0.27.1 slam_0.1-47 GenomeInfoDbData_1.2.2
[25] KMsurv_0.1-5 farver_2.0.3 bit64_0.9-7
[28] rprojroot_1.3-2 vctrs_0.3.1 generics_0.0.2
[31] TH.data_1.0-10 xfun_0.15 BiocFileCache_1.10.2
[34] sets_1.0-18 R6_2.4.1 doParallel_1.0.15
[37] clue_0.3-57 bitops_1.0-6 fgsea_1.12.0
[40] assertthat_0.2.1 promises_1.1.1 scales_1.1.1
[43] multcomp_1.4-13 gtable_0.3.0 affy_1.64.0
[46] sandwich_2.5-1 workflowr_1.6.2 rlang_0.4.7
[49] mzR_2.20.0 GlobalOptions_0.1.2 splines_3.6.0
[52] rstatix_0.6.0 impute_1.60.0 hexbin_1.28.1
[55] broom_0.7.0 modelr_0.1.8 BiocManager_1.30.10
[58] yaml_2.2.1 abind_1.4-5 crosstalk_1.1.0.1
[61] backports_1.1.8 httpuv_1.5.4 tools_3.6.0
[64] relations_0.6-9 affyio_1.56.0 ellipsis_0.3.1
[67] gplots_3.0.4 RColorBrewer_1.1-2 MSnbase_2.12.0
[70] Rcpp_1.0.5 plyr_1.8.6 visNetwork_2.0.9
[73] progress_1.2.2 zlibbioc_1.32.0 RCurl_1.98-1.2
[76] prettyunits_1.1.1 openssl_1.4.2 ggpubr_0.4.0
[79] GetoptLong_1.0.2 zoo_1.8-8 haven_2.3.1
[82] cluster_2.1.0 exactRankTests_0.8-31 fs_1.4.2
[85] magrittr_1.5 data.table_1.12.8 openxlsx_4.1.5
[88] circlize_0.4.10 reprex_0.3.0 survminer_0.4.7
[91] pcaMethods_1.78.0 mvtnorm_1.1-1 ProtGenerics_1.18.0
[94] hms_0.5.3 shinyjs_1.1 mime_0.9
[97] evaluate_0.14 xtable_1.8-4 XML_3.98-1.20
[100] rio_0.5.16 readxl_1.3.1 gridExtra_2.3
[103] shape_1.4.4 compiler_3.6.0 KernSmooth_2.23-17
[106] ncdf4_1.17 crayon_1.3.4 htmltools_0.5.0
[109] mgcv_1.8-31 later_1.1.0.1 lubridate_1.7.9
[112] DBI_1.1.0 dbplyr_1.4.4 ComplexHeatmap_2.2.0
[115] rappdirs_0.3.1 MASS_7.3-51.6 tmvtnorm_1.4-10
[118] Matrix_1.2-18 car_3.0-8 cli_2.0.2
[121] imputeLCMD_2.0 marray_1.64.0 gdata_2.18.0
[124] igraph_1.2.5 pkgconfig_2.0.3 km.ci_0.5-2
[127] foreign_0.8-71 piano_2.2.0 xml2_1.3.2
[130] MALDIquant_1.19.3 foreach_1.5.0 XVector_0.26.0
[133] drc_3.0-1 rvest_0.3.5 digest_0.6.25
[136] rmarkdown_2.3 cellranger_1.1.0 fastmatch_1.1-0
[139] survMisc_0.5.5 curl_4.3 shiny_1.5.0
[142] gtools_3.8.2 rjson_0.2.20 nlme_3.1-148
[145] lifecycle_0.2.0 jsonlite_1.7.0 carData_3.0-4
[148] fansi_0.4.1 askpass_1.1 pillar_1.4.6
[151] lattice_0.20-41 httr_1.4.1 fastmap_1.0.1
[154] plotrix_3.7-8 survival_3.2-3 glue_1.4.1
[157] zip_2.0.4 png_0.1-7 iterators_1.0.12
[160] bit_1.1-15.2 stringi_1.4.6 blob_1.2.1
[163] caTools_1.18.0 memoise_1.1.0