Quality control of the proteomic data from timsTOF machine
Junyan Lu
2020-02-27
Last updated: 2020-03-10
Checks: 7 0
Knit directory: Proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/compareProteomicsRNAseq_cache/
Ignored: analysis/correlateCLLPD_cache/
Ignored: analysis/correlateGenomic_cache/
Ignored: analysis/correlateGenomic_removePC_cache/
Ignored: analysis/correlateMIR_cache/
Ignored: analysis/correlateMethylationCluster_cache/
Ignored: analysis/predictOutcome_cache/
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: code/utils.R
Untracked: data/190909_CLL_prot_abund_med_norm.tsv
Untracked: data/190909_CLL_prot_abund_no_norm.tsv
Untracked: data/20190423_Proteom_submitted_samples_bereinigt.xlsx
Untracked: data/20191025_Proteom_submitted_samples_final.xlsx
Untracked: data/LUMOS/
Untracked: data/LUMOS_peptides/
Untracked: data/LUMOS_protAnnotation.csv
Untracked: data/SampleAnnotation_cleaned.xlsx
Untracked: data/facTab_IC50atLeast3New.RData
Untracked: data/gmts/
Untracked: data/mapEnsemble.txt
Untracked: data/mapSymbol.txt
Untracked: data/pyprophet_export_aligned.csv
Untracked: data/timsTOF_protAnnotation.csv
Untracked: output/LUMOS_processed.RData
Untracked: output/proteomic_LUMOS_20200227.RData
Untracked: output/proteomic_timsTOF_20200227.RData
Untracked: output/timsTOF_processed.RData
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 46534c2 | Junyan Lu | 2020-02-27 | Build site. |
| Rmd | 2b8852e | Junyan Lu | 2020-02-27 | wflow_publish(list.files(“./”, pattern = “Rmd”)) |
Test association with RNA expression
Dimension of the inputed data
dim(protCLL)[1] 3329 49Process both datasets
colnames(dds) <- dds$PatID
dds <- estimateSizeFactors(dds)
sampleOverlap <- intersect(colnames(protCLL), colnames(dds))
geneOverlap <- intersect(rowData(protCLL)$ensembl_gene_id, rownames(dds))
ddsSub <- dds[geneOverlap, sampleOverlap]
protSub <- protCLL[match(geneOverlap, rowData(protCLL)$ensembl_gene_id), sampleOverlap]
#how many gene don't have RNA expression at all?
noExp <- rowSums(counts(ddsSub)) == 0
sum(noExp)[1] 11#remove those genes in both datasets
ddsSub <- ddsSub[!noExp,]
protSub <- protSub[!noExp,]
#remove proteins with duplicated identifiers
protSub <- protSub[!duplicated(rowData(protSub)$name)]
geneOverlap <- intersect(rowData(protSub)$ensembl_gene_id, rownames(ddsSub))
ddsSub.vst <- varianceStabilizingTransformation(ddsSub)Calculate correlations between protein abundance and RNA expression
rnaMat <- assay(ddsSub.vst)
proMat.raw <- assays(protSub)[["count"]]
proMat.qrilc <- assays(protSub)[["QRILC"]]
rownames(proMat.qrilc) <- rowData(protSub)$ensembl_gene_id
rownames(proMat.raw) <- rowData(protSub)$ensembl_gene_id
corTab <- lapply(geneOverlap, function(n) {
rna <- rnaMat[n,]
pro.q <- proMat.qrilc[n,]
pro.raw <- proMat.raw[n,]
res.q <- cor.test(rna, pro.q)
res.raw <- cor.test(rna, pro.raw, use = "pairwise.complete.obs")
tibble(id = n, impute=c("No Imputation","QRILC"),
p = c(res.raw$p.value, res.q$p.value),
coef = c(res.raw$estimate, res.q$estimate))
}) %>% bind_rows() %>%
arrange(desc(coef)) %>% mutate(p.adj = p.adjust(p, method = "BH"),
symbol = rowData(dds[id,])$symbol)Plot the distribution of correlation coefficient
ggplot(corTab, aes(x=coef, fill = impute)) + geom_histogram(position = "identity", col = "grey50", alpha =0.3, bins =100) +
geom_vline(xintercept = 0, col = "red", linetype = "dashed")Warning: Removed 2 rows containing non-finite values (stat_bin).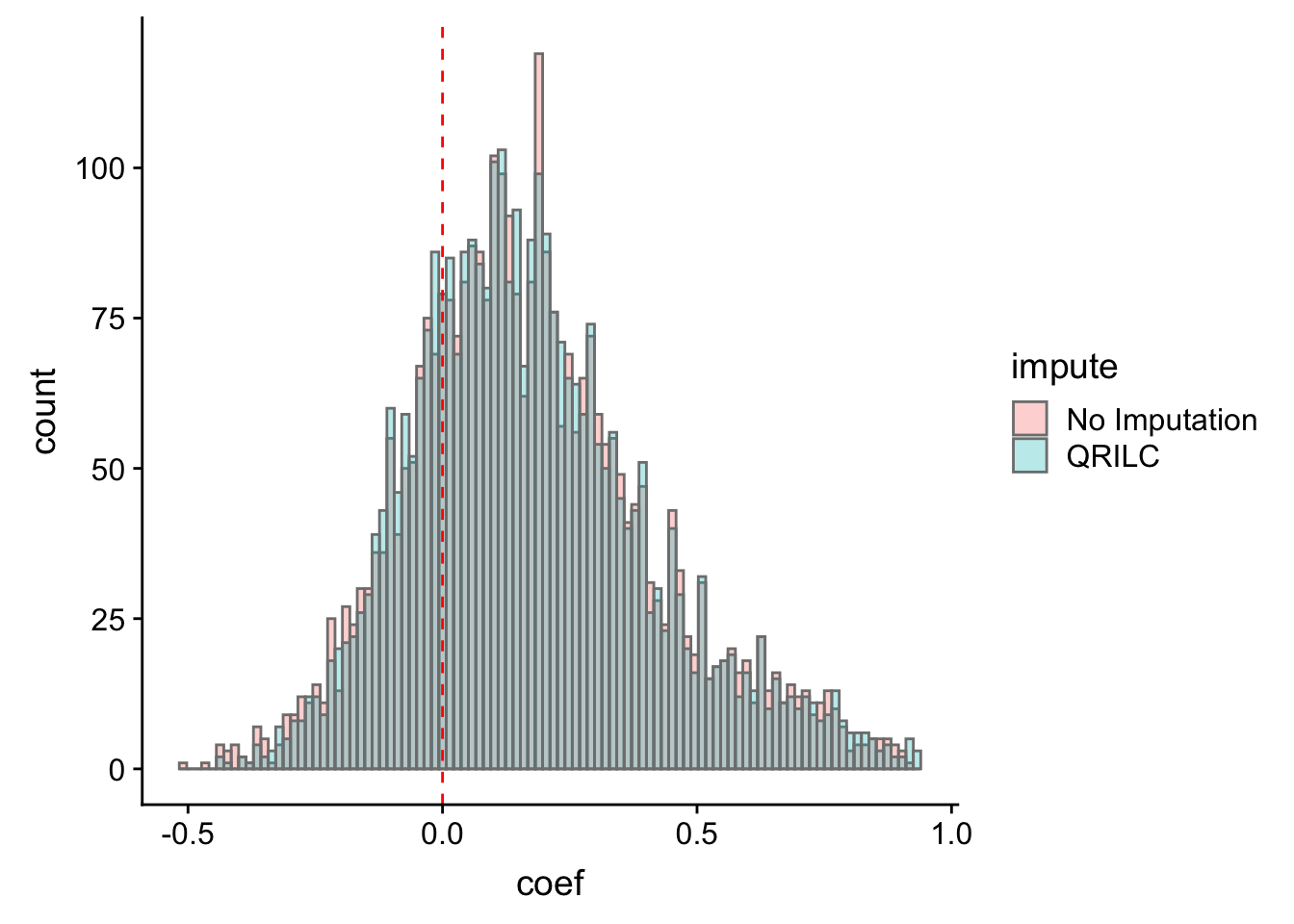
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Most of the correlations are positive, which is reasonable.
Number of significant positive and negative correlations (10% FDR)
sigTab <- corTab %>% filter(p.adj < 0.1) %>% mutate(direction = ifelse(coef > 0, "positive", "negative")) %>%
group_by(impute, direction) %>% summarise(number = length(id)) %>% ungroup() %>%
mutate(ratio = format(number/length(geneOverlap), digits = 2)) %>% arrange(number)
DT::datatable(sigTab)Number of significant correlations VS FDR cut-off
plotTab <- lapply(seq(0,0.1, length.out = 100), function(fdr) {
filTab <- dplyr::filter(corTab, p.adj < fdr, coef > 0) %>%
group_by(impute) %>% summarise(n = length(id)) %>% mutate(fdr = fdr)
}) %>% bind_rows()
ggplot(plotTab, aes(x=fdr, y = n, col = impute))+ geom_line() +
ylab("Number of significant correlations") +
xlab("FDR cut-off")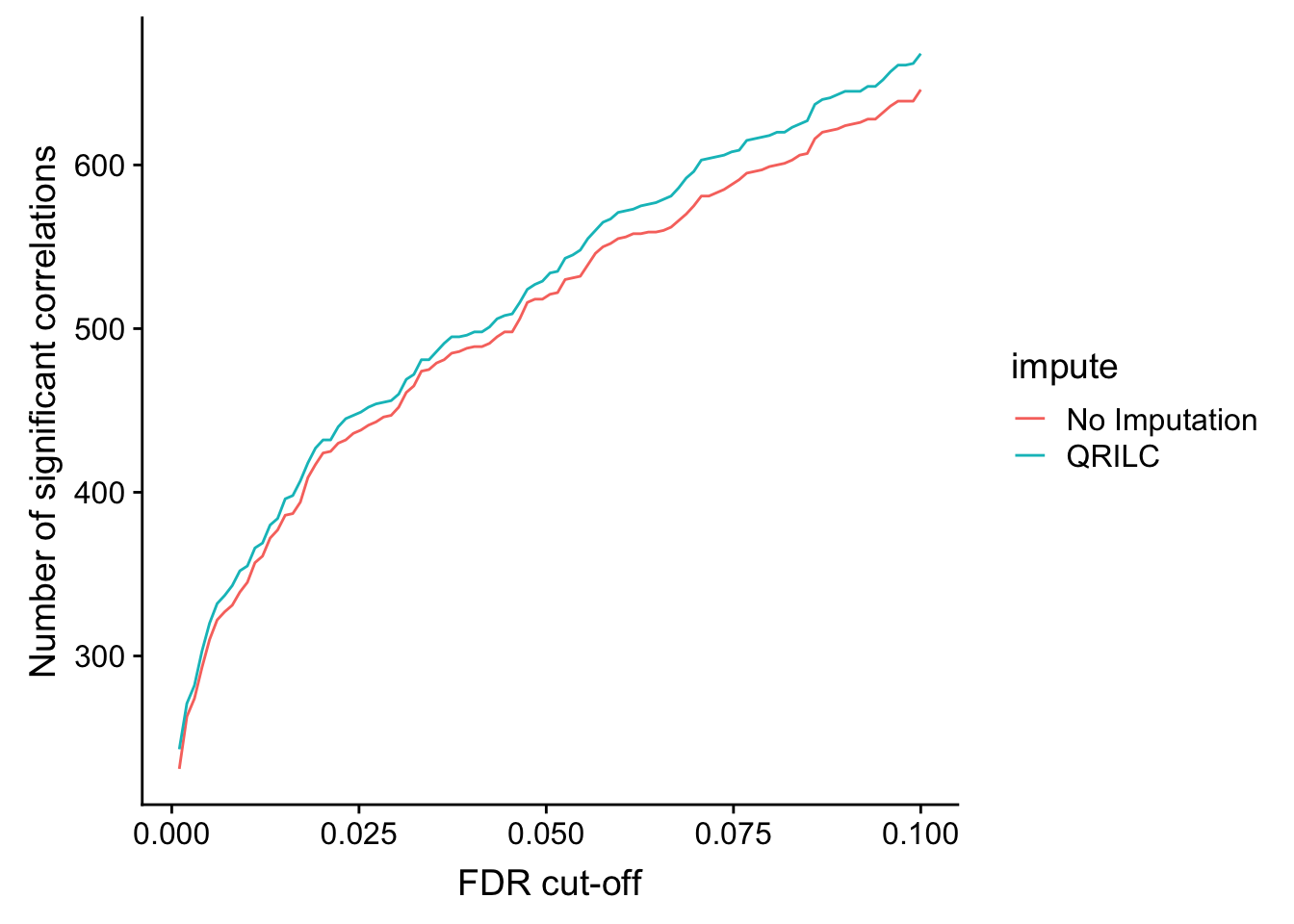
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
List of proteins that significantly correlated with RNA expression (10 %FDR, no imputation)
sigTab <- filter(corTab, p.adj < 0.1, impute == "No Imputation") %>% mutate_if(is.numeric, format, digits=2)
DT::datatable(sigTab)List of proteins that significantly correlated with RNA expression (10 %FDR, QRILC imputed)
sigTab <- filter(corTab, p.adj < 0.1, impute == "QRILC") %>% mutate_if(is.numeric, format, digits=2)
DT::datatable(sigTab)Correlation plot of top 9 most correlated protein-rna pairs
plotList <- lapply(sigTab$id[1:9], function(n) {
plotTab <- tibble(pro = proMat.qrilc[n,], gene = rnaMat[n,])
symbol <- filter(sigTab, id == n)$symbol
ggplot(plotTab, aes(x=pro, y=gene)) + geom_point() + geom_smooth(method = "lm") +
ggtitle(symbol) + ylab("RNA expression") + xlab("Protein abundance")
})
cowplot::plot_grid(plotlist = plotList, ncol =3)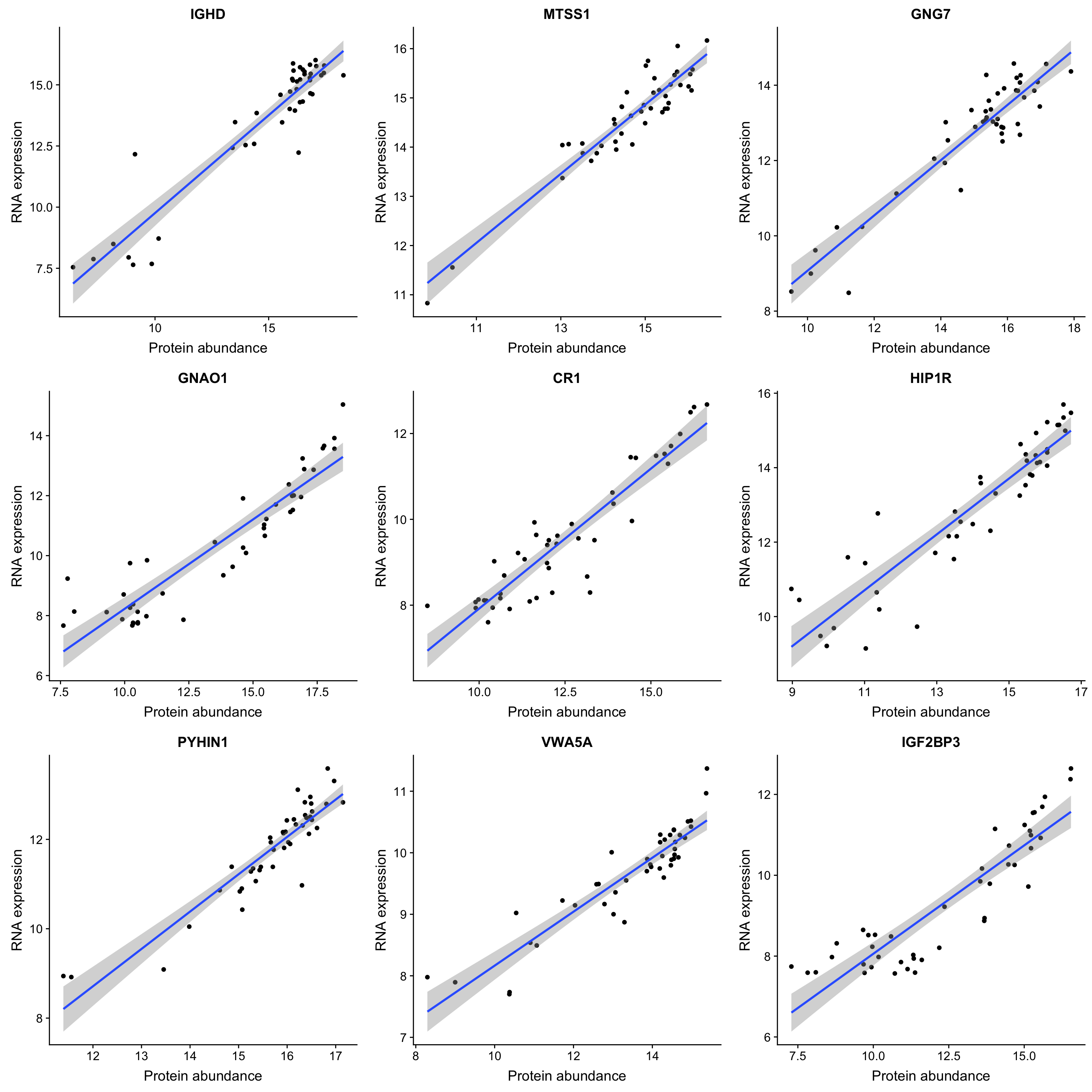
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Assocation with technical factors
Are thechnical variables confounded with major genomic variabes?
# A tibble: 3 x 4
genomic technical pval p.adj
<chr> <chr> <dbl> <dbl>
1 IGHV.status processDate 0.0219 0.480
2 SF3B1 freeThawCycle 0.0229 0.480
3 del13q operator 0.0494 0.692No Significant assocations
Association between technical variables and priciple components of protein expression
plotMat <- assays(protCLL)[["QRILC"]]
pcRes <- prcomp(t(plotMat), center =TRUE, scale. = FALSE)$x
testRes <- lapply(colnames(pcRes), function(pc) {
lapply(colnames(techTab), function(tech) {
pcVar <- pcRes[,pc]
techVar <- techTab[[tech]]
res <- summary(aov(pcVar ~ techVar))
p <- res[[1]][1,4]
tibble(component = pc, technical = tech, pval = p)
}) %>% bind_rows()
}) %>% bind_rows() %>% mutate(p.adj = p.adjust(pval, method = "BH")) %>%
arrange(pval)
filter(testRes, p.adj < 0.1)# A tibble: 10 x 4
component technical pval p.adj
<chr> <chr> <dbl> <dbl>
1 PC31 proteinConc 0.0000391 0.0115
2 PC47 proteinConc 0.000124 0.0158
3 PC41 proteinConc 0.000162 0.0158
4 PC45 proteinConc 0.000806 0.0593
5 PC27 proteinConc 0.00143 0.0843
6 PC25 proteinConc 0.00216 0.0991
7 PC12 proteinConc 0.00237 0.0991
8 PC48 proteinConc 0.00271 0.0991
9 PC18 freeThawCycle 0.00336 0.0991
10 PC49 proteinConc 0.00337 0.0991There are some principle components correlated with technical variables. But the PCs are not top PCs, suggesting the know technical factor do not have impact on the major trends of the dataset.
Associations between technical variables and individual protein expressions
Association test
techTab <- colData(protCLL)[,c("operator", "viability","batch","processDate","proteinConc","freeThawCycle")] %>%
data.frame() %>%rownames_to_column("patID") %>% as_tibble() %>% mutate(processDate = as.character(processDate)) %>%
mutate_if(is.character, as.factor)%>% mutate_at(vars(-patID), as.numeric)
testTab <- assays(protCLL)[["QRILC"]] %>% data.frame() %>%
rownames_to_column("id") %>% mutate(name = rowData(protCLL)[id,]$hgnc_symbol) %>%
gather(key = "patID", value = "expr", -id, -name) %>%
left_join(techTab, by ="patID") %>% gather(key = technical, value = value, -id, -name, -patID, -expr)Warning: Column `patID` joining character vector and factor, coercing into
character vectortestRes <- filter(testTab, !is.na(value)) %>%
group_by(name, technical) %>% nest() %>%
mutate(m = map(data, ~lm(expr~value,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% filter(term=="value") %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))
sumTab <- filter(testRes, p.adj < 0.1) %>% group_by(technical) %>% summarise(n=length(name))
ggplot(sumTab, aes(x=technical, y = n)) + geom_bar(stat = "identity") + coord_flip() +
xlab("") + ylab("Number of significantly associated proteins")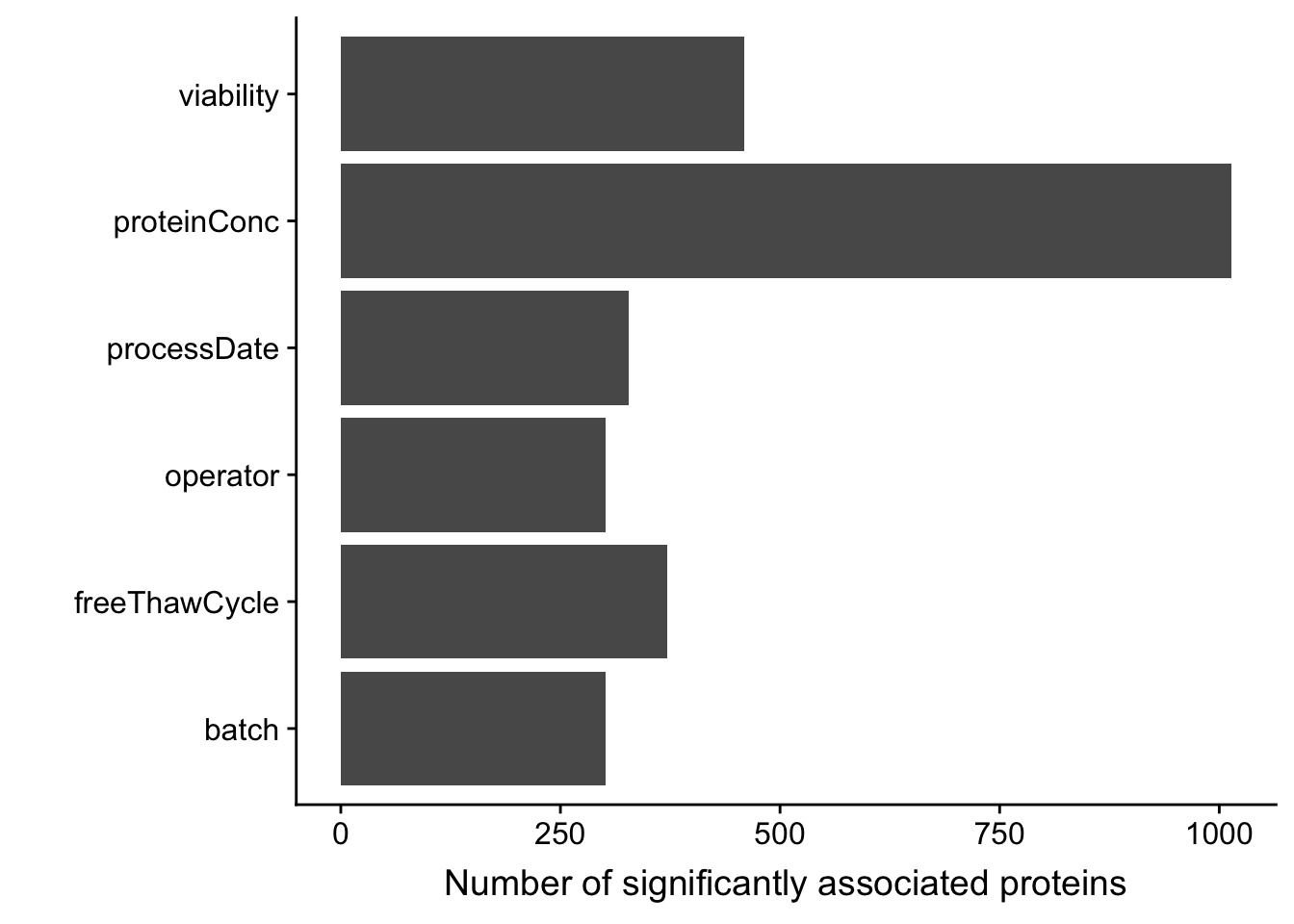
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Assocation P value histogram for each technical factor
ggplot(testRes, aes(x=p.value)) + geom_histogram() + facet_wrap(~technical) +xlim(0,1)`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.Warning: Removed 12 rows containing missing values (geom_bar).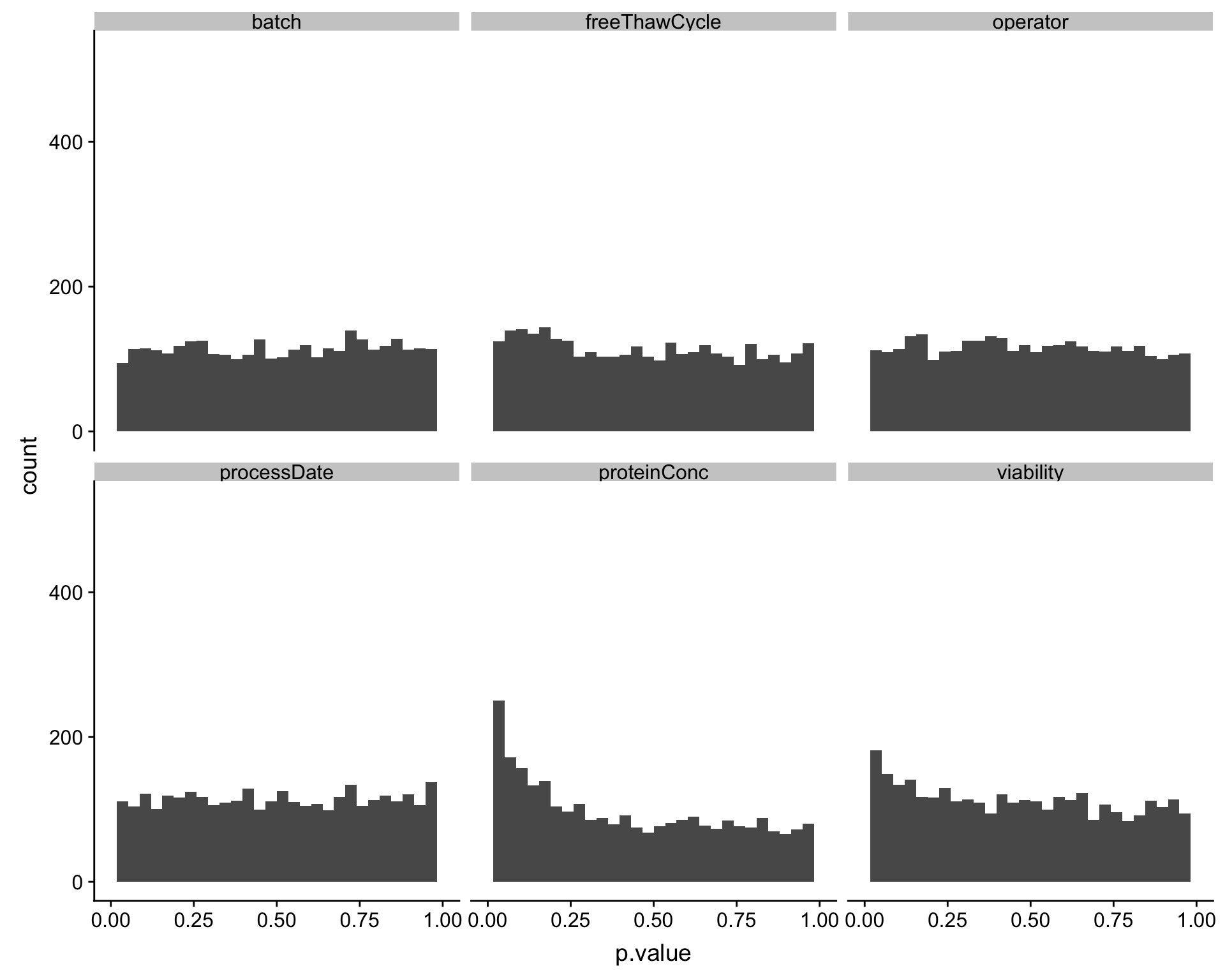
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Also based on the p-value histogram, only overall protein concentration may have potential impact on protein abundance detection.
Will the correlation with RNA expression improve if we adjust for total protein concentration?
proteinConc <- techTab[match(colnames(proMat.qrilc), techTab$patID),]$proteinConc
corTab <- lapply(geneOverlap, function(n) {
rna <- rnaMat[n,]
pro.q <- proMat.qrilc[n,]
p.single <- anova(lm(rna ~ pro.q))$`Pr(>F)`[1]
p.multi <- car::Anova(lm(rna ~ pro.q + proteinConc))$`Pr(>F)`[1]
tibble(name = n, corrected = c("no","yes"),
p = c(p.single, p.multi))
}) %>% bind_rows() %>% mutate(p.adj = p.adjust(p, method = "BH")) %>% arrange(p)Number of significant correlations VS FDR cut-off
plotTab <- lapply(seq(0,0.1, length.out = 100), function(fdr) {
filTab <- dplyr::filter(corTab, p.adj < fdr) %>%
group_by(corrected) %>% summarise(n = length(name)) %>% mutate(fdr = fdr)
}) %>% bind_rows()
ggplot(plotTab, aes(x=fdr, y = n, col = corrected))+ geom_line() +
ylab("Number of significant correlations") +
xlab("FDR cut-off")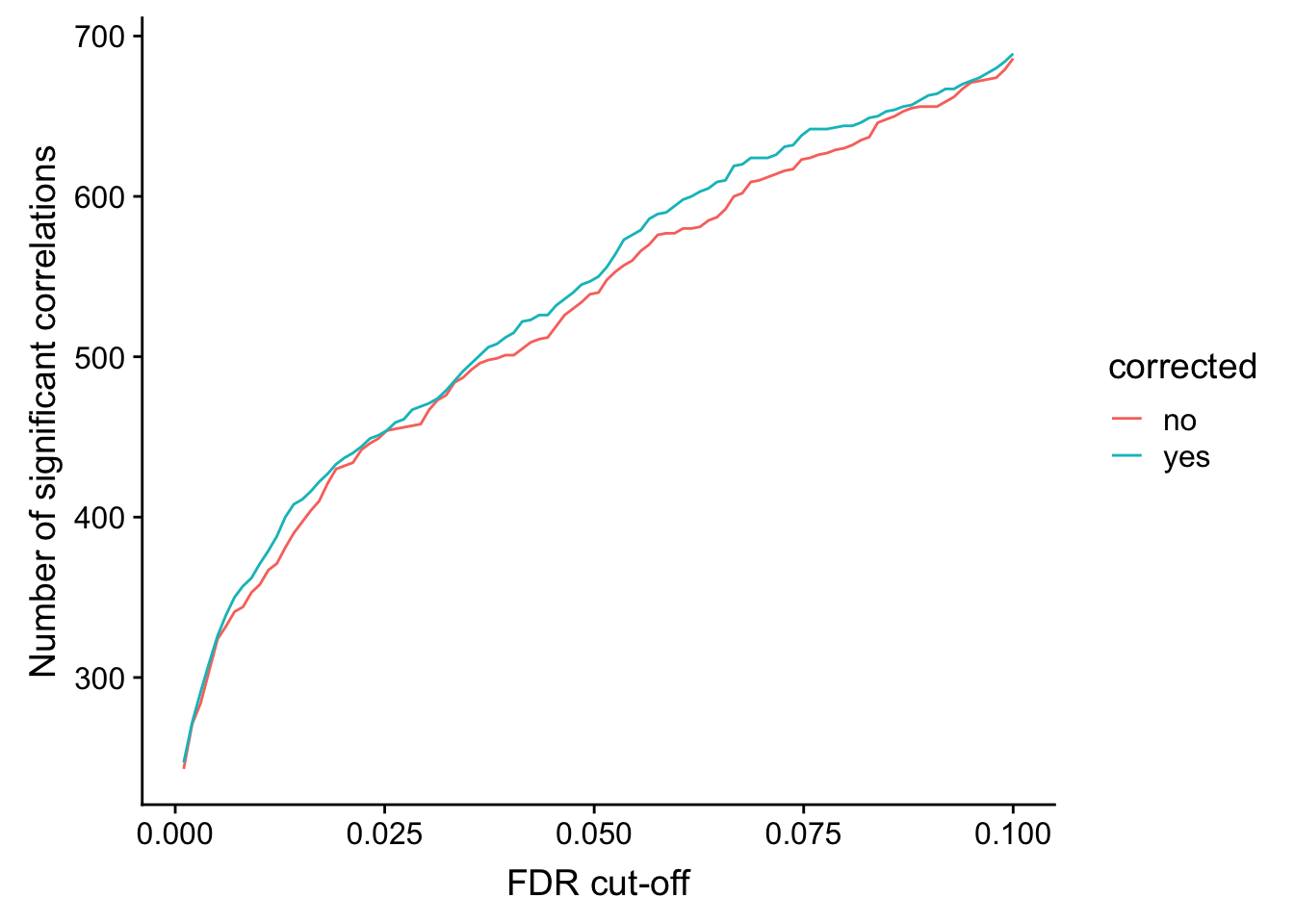
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Seems to improve the correlation a little, but not much. We can include this factor in association test later.
Check data structure
Hierarchical clustering
plotMat <- assays(protCLL)[["QRILC"]]
colAnno <- colData(protCLL)[,c("gender","IGHV.status","trisomy12")] %>%
data.frame()
plotMat <- jyluMisc::mscale(plotMat, censor = 6)
pheatmap(plotMat, scale = "none", annotation_col = colAnno, clustering_method = "ward.D2",
show_rownames = FALSE, color = colorRampPalette(c("navy","white","firebrick"))(100),
breaks = seq(-6,6, length.out = 101))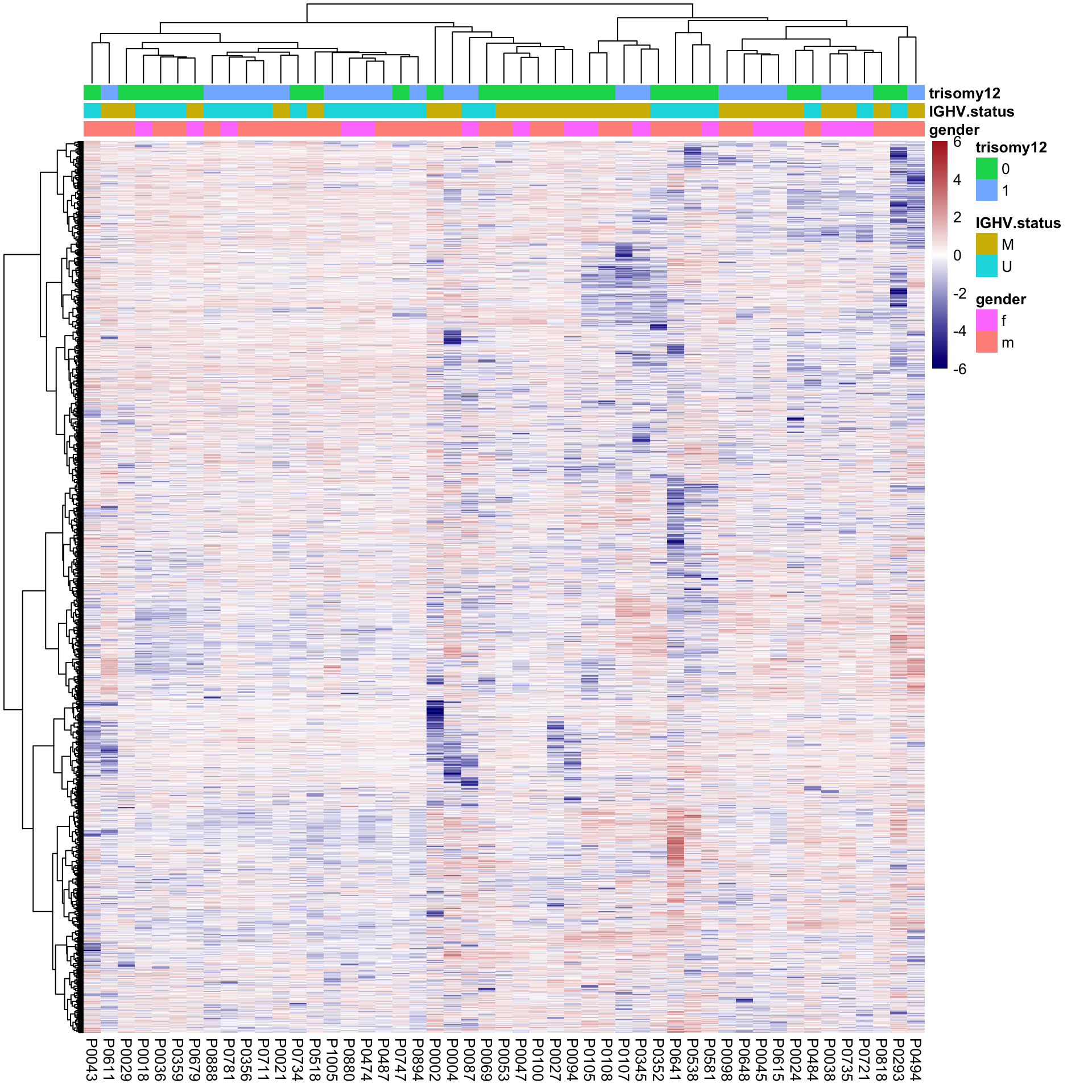
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
No clear separation can be observed
PCA
pcOut <- prcomp(t(plotMat), center =TRUE, scale. = FALSE)
pcRes <- pcOut$x
eigs <- pcOut$sdev^2
varExp <- structure(eigs/sum(eigs),names = colnames(pcRes))
plotTab <- pcRes[,1:2] %>% data.frame() %>% cbind(colAnno[rownames(.),]) %>%
rownames_to_column("patID") %>% as_tibble()
ggplot(plotTab, aes(x=PC1, y=PC2, col = IGHV.status, shape = trisomy12)) + geom_point(size=4) +
xlab(sprintf("PC1 (%1.2f%%)",varExp[["PC1"]]*100)) +
ylab(sprintf("PC2 (%1.2f%%)",varExp[["PC2"]]*100))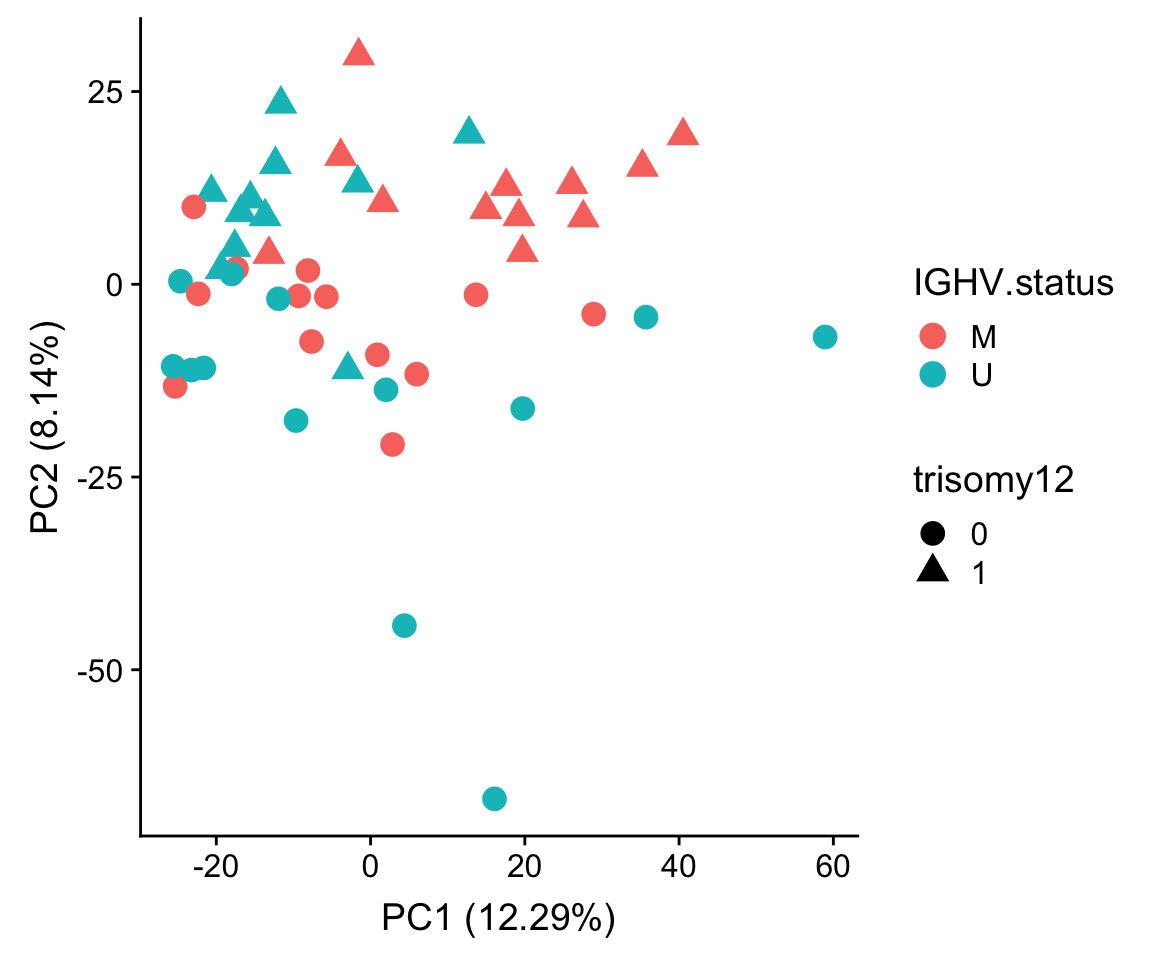
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
PC2 separates trisomy12
Correlation PCs with trisomy12 and IGHV status
corTab <- lapply(colnames(pcRes), function(pc) {
ighvCor <- t.test(pcRes[,pc] ~ colAnno$IGHV.status)
tri12Cor <- t.test(pcRes[,pc] ~ colAnno$trisomy12)
tibble(PC = pc,
feature=c("IGHV", "trisomy12"),
p = c(ighvCor$p.value, tri12Cor$p.value))
}) %>% bind_rows() %>% mutate(p.adj = p.adjust(p, method = "BH")) %>%
filter(p <= 0.05) %>% arrange(p)
corTab# A tibble: 6 x 4
PC feature p p.adj
<chr> <chr> <dbl> <dbl>
1 PC2 trisomy12 0.000000348 0.0000342
2 PC6 IGHV 0.0000912 0.00447
3 PC5 trisomy12 0.00197 0.0642
4 PC4 IGHV 0.0295 0.598
5 PC3 IGHV 0.0305 0.598
6 PC49 IGHV 0.0470 0.682 PC6 is for IGHV
PCA plot using PC2 and PC7
plotTab <- pcRes[,1:10] %>% data.frame() %>% cbind(colAnno[rownames(.),]) %>%
rownames_to_column("patID") %>% as_tibble()
ggplot(plotTab, aes(x=PC2, y=PC6, col = IGHV.status, shape = trisomy12, label = patID)) + geom_point() + ggrepel::geom_text_repel() +
xlab(sprintf("PC2 (%1.2f%%)",varExp[["PC2"]]*100)) +
ylab(sprintf("PC6 (%1.2f%%)",varExp[["PC6"]]*100))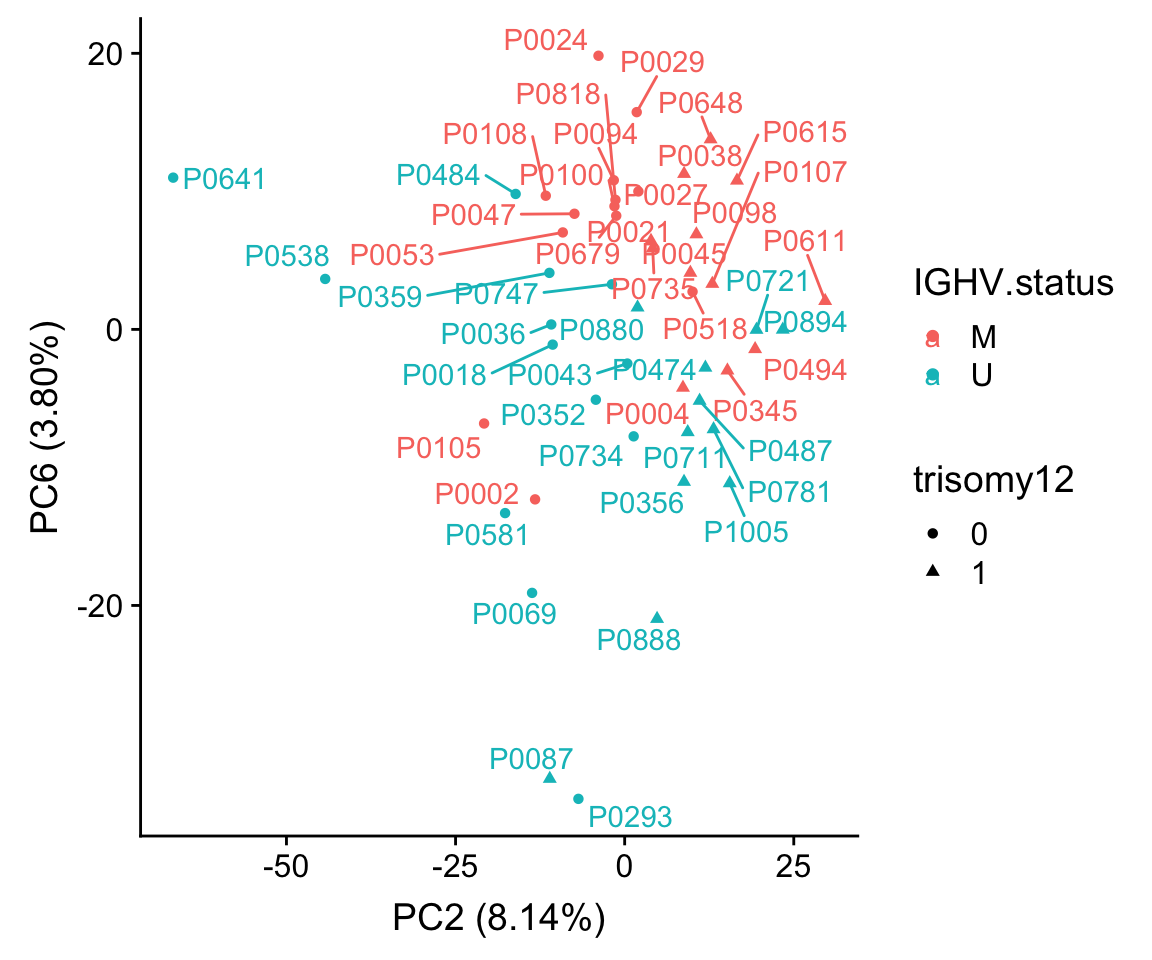
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Using PC3 and PC4 better seperates IGHV and trisomy12.
Biological annotation of PC1
Annotate PC1
Correlation test
Assocation test
proMat <- assays(protCLL)[["QRILC"]]
pc <- pcRes[,1][colnames(proMat)]
designMat <- model.matrix(~1+pc)
fit <- lmFit(proMat, designMat)
fit2 <- eBayes(fit)
corRes.pc1 <- topTable(fit2, number ="all", adjust.method = "BH", coef = "pc") %>% rownames_to_column("id") %>%
mutate(symbol = rowData(protCLL[id,])$hgnc_symbol)Number of significant associations (10% FDR)
hist(corRes.pc1$P.Value,breaks=100)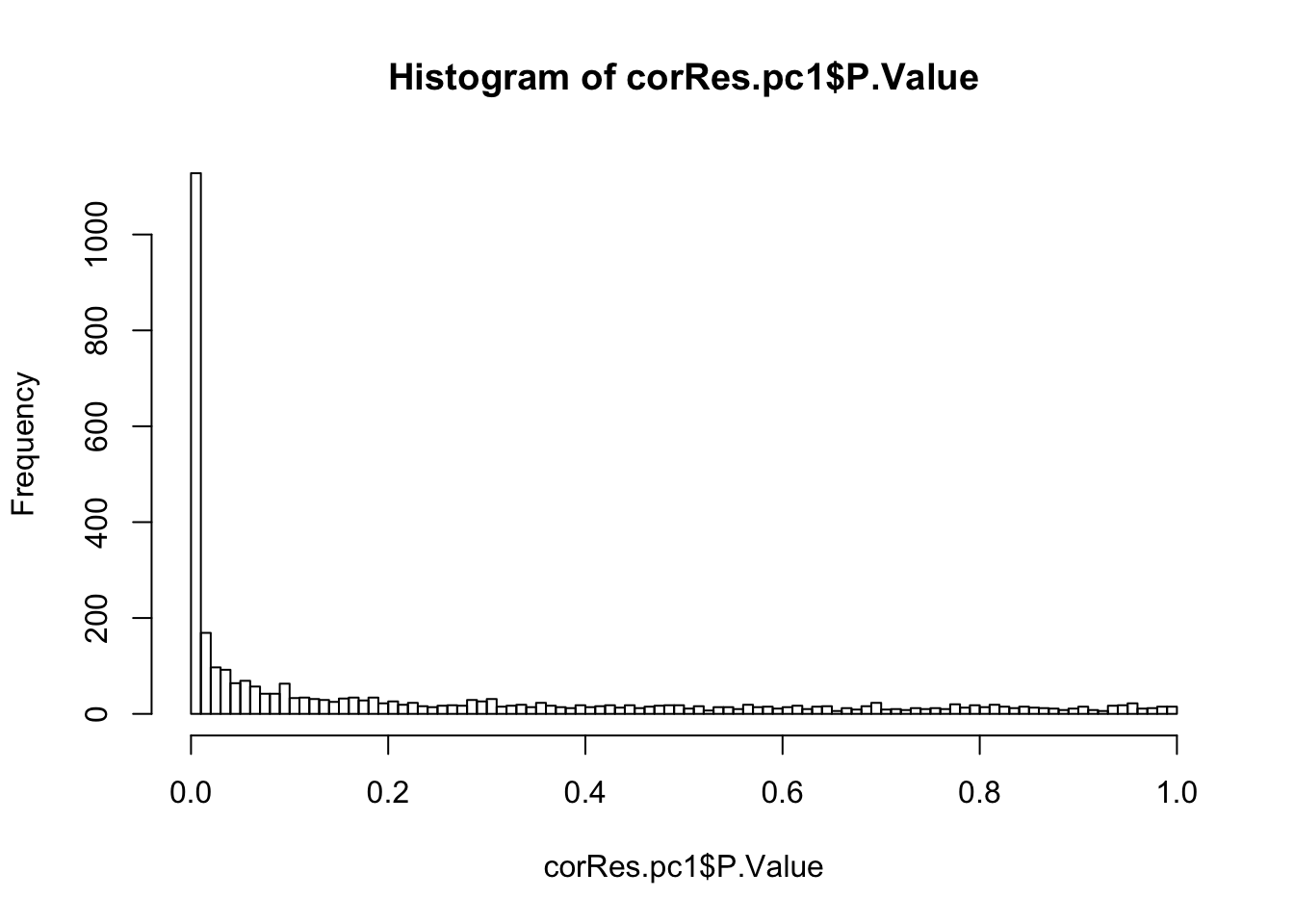
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Table of significant associations (5% FDR)
resTab.sig <- filter(corRes.pc1, adj.P.Val < 0.05) %>%
select(symbol, id,logFC, P.Value, adj.P.Val) %>%
arrange(P.Value)
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2, format= "e") %>%
DT::datatable()Heatmap of associated genes
colAnno <- tibble(patID = colnames(proMat), PC1 = pcRes[colnames(proMat),1]) %>%
arrange(PC1) %>% data.frame() %>% column_to_rownames("patID")
plotMat <- proMat[resTab.sig$id[1:100],rownames(colAnno)]
plotMat <- jyluMisc::mscale(plotMat, censor = 6)
pheatmap(plotMat, scale = "none", annotation_col = colAnno, clustering_method = "ward.D2",
cluster_cols = FALSE,
labels_row = resTab.sig$symbol[1:100], color = colorRampPalette(c("navy","white","firebrick"))(100),
breaks = seq(-6,6, length.out = 101))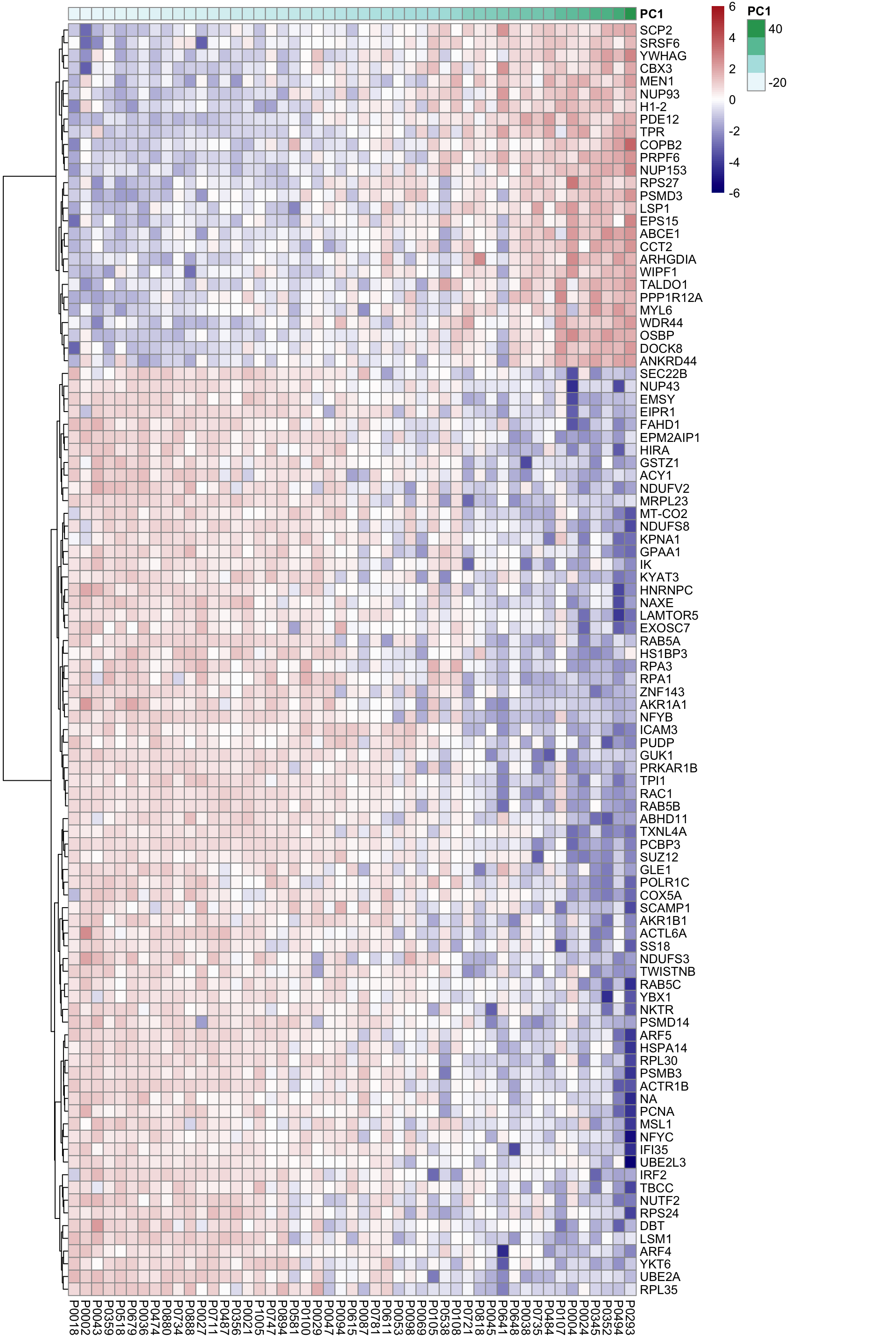
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Enrichment using Camera
Hallmarks
gmts = list(H= "../data/gmts/h.all.v6.2.symbols.gmt",
C6 = "../data/gmts/c6.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
res <- runCamera(proMat, designMat, gmts$H, id = rowData(protCLL[rownames(proMat),])$hgnc_symbol)
res$enrichPlot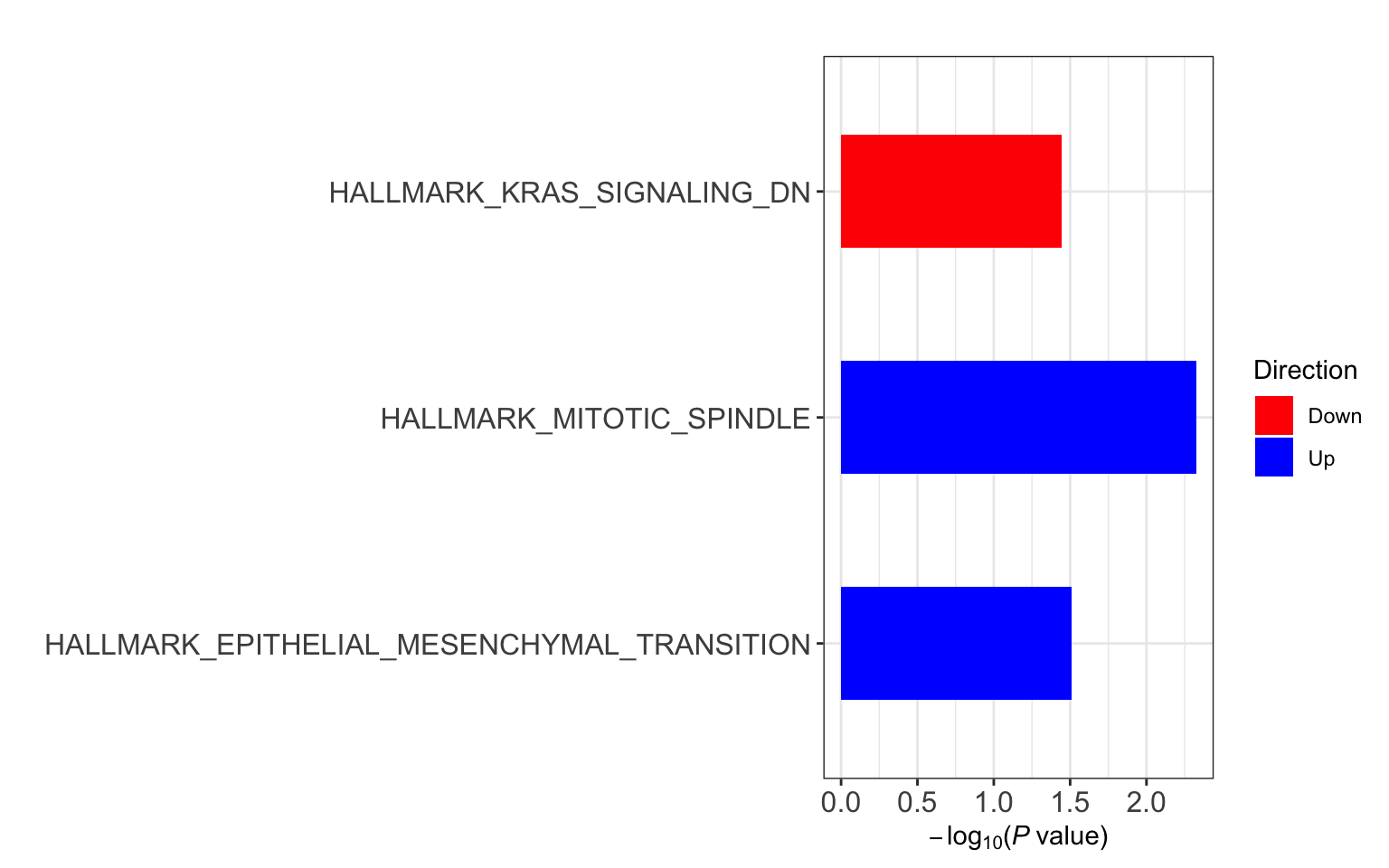
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
C6
res <- runCamera(proMat, designMat, gmts$C6, id = rowData(protCLL[rownames(proMat),])$hgnc_symbol)
res$enrichPlot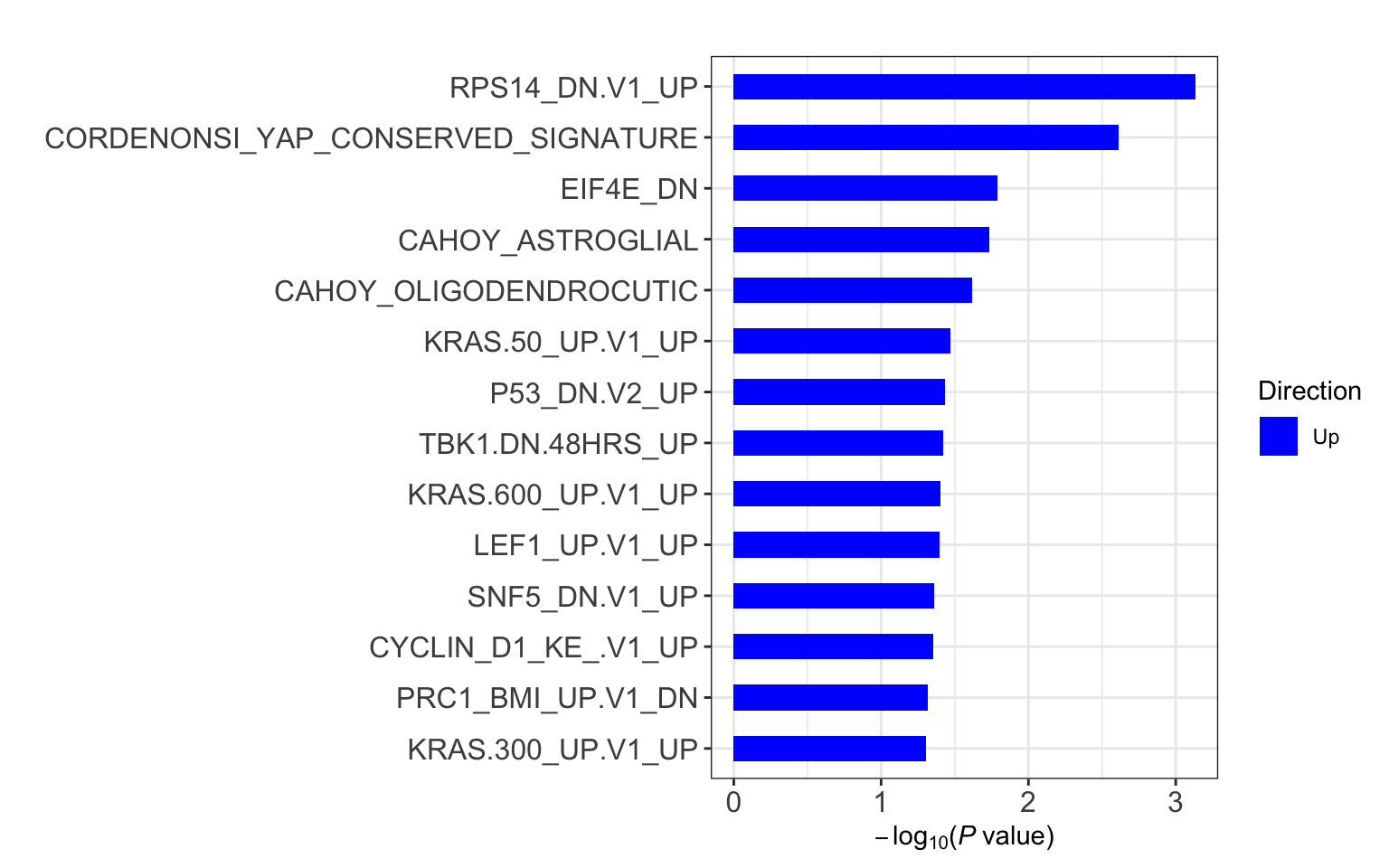
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
KEGG
res <- runCamera(proMat, designMat, gmts$KEGG, id = rowData(protCLL[rownames(proMat),])$hgnc_symbol)
res$enrichPlot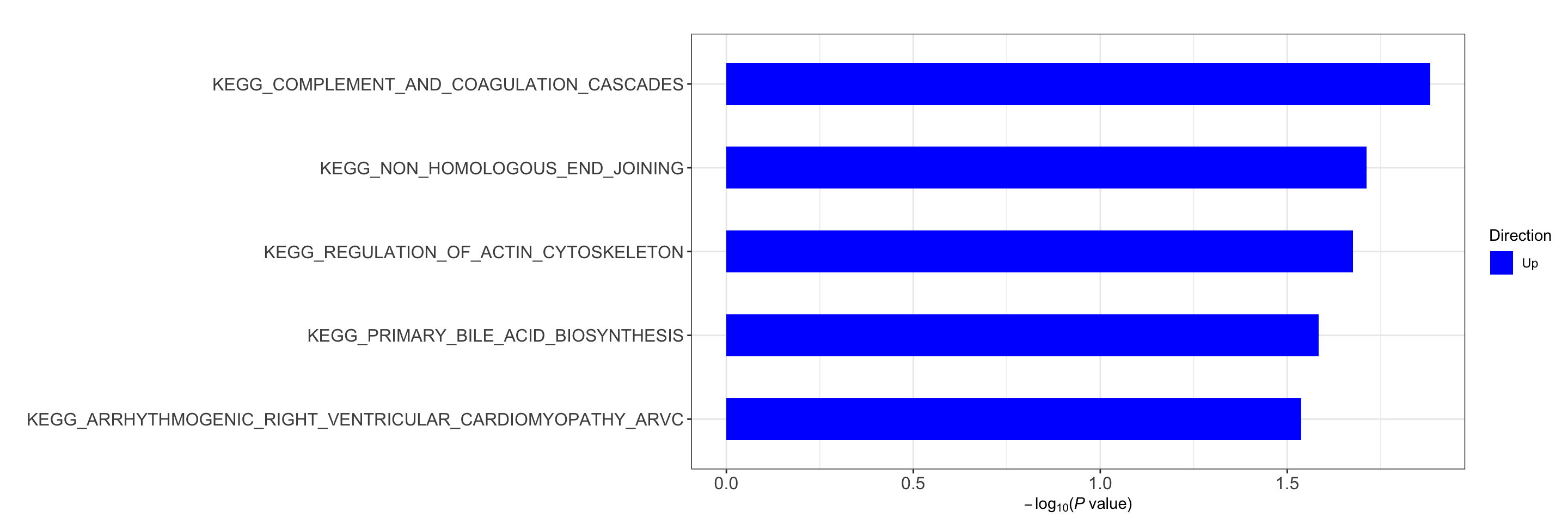
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Association with lymphocyte count (potential contamination?)
load("~/CLLproject_jlu/ShinyApps/sampleTimeline//timeline.RData")
plotTab <- pcRes[,1:9] %>% data.frame() %>%
rownames_to_column("patID") %>% as_tibble() %>%
mutate(sampleID = protCLL[,patID]$sampleID) %>%
mutate(lymCount = sampleTab[match(sampleID, sampleTab$sampleID),]$leukCount) %>%
gather(key = "pc", value = "val",-patID,-sampleID,-lymCount)
ggplot(plotTab, aes(x=val, y=lymCount)) + geom_point() + geom_smooth(method = "lm") +
facet_wrap(~pc, ncol =3, scale = "free")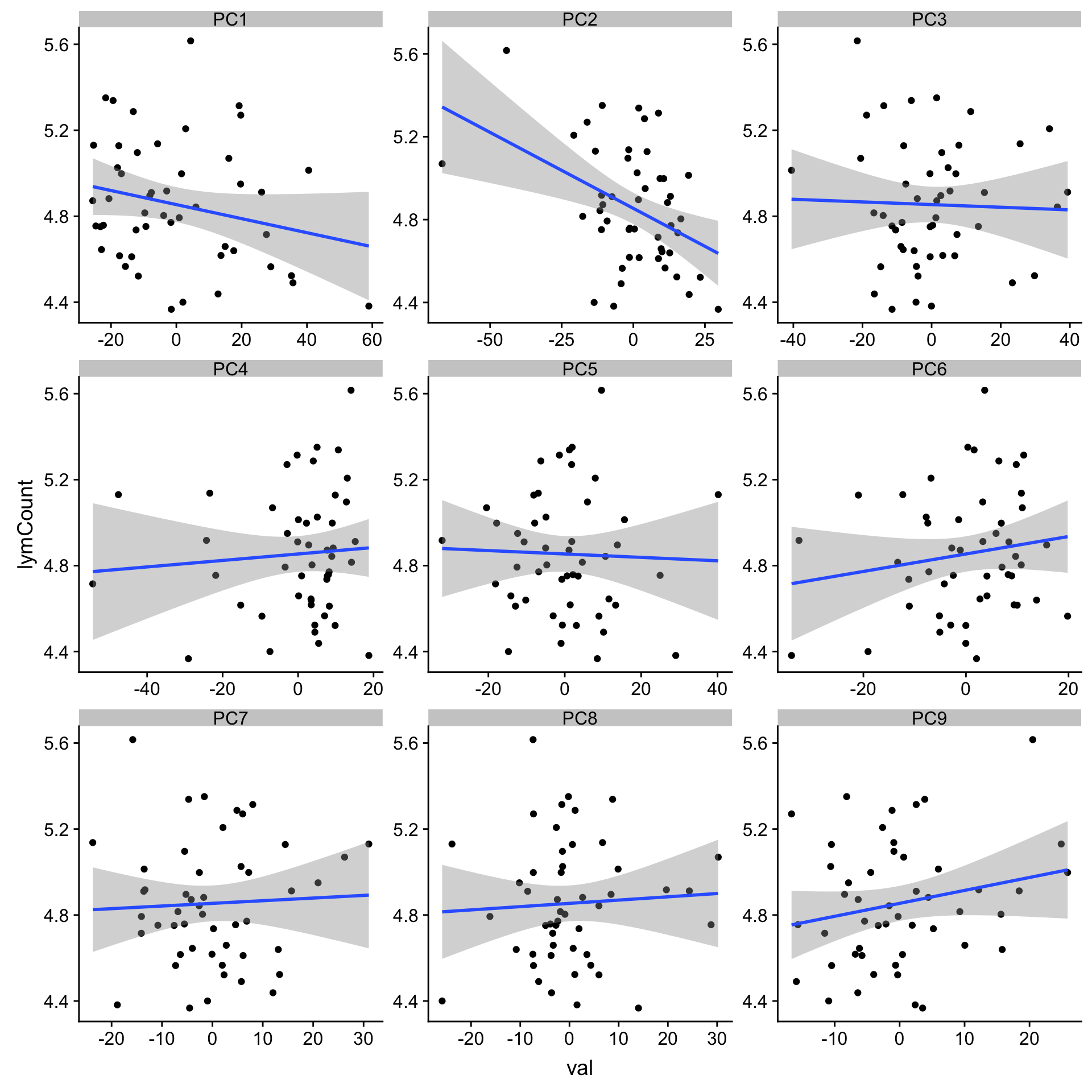
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
corRes <- plotTab %>% group_by(pc) %>% nest() %>%
mutate(m= map(data, ~cor.test(~ val+ lymCount,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(pc, estimate, p.value) %>%
arrange(p.value)
corRes# A tibble: 9 x 3
# Groups: pc [9]
pc estimate p.value
<chr> <dbl> <dbl>
1 PC2 -0.420 0.00265
2 PC1 -0.230 0.111
3 PC9 0.211 0.145
4 PC6 0.159 0.275
5 PC4 0.0781 0.594
6 PC8 0.0571 0.697
7 PC7 0.0482 0.742
8 PC5 -0.0356 0.808
9 PC3 -0.0338 0.818 Association wit %Lymphocyte PB
library(DBI)
con <- dbConnect(RPostgreSQL::PostgreSQL(),
dbname = "tumorbank",
host = "huber-vm01.embl.de",
user = "admin",
password = "bloodcancertumorbank")
PBtab <- tbl(con, "patient") %>%
left_join(tbl(con, "sample"), by = c(patid = "smppatidref")) %>%
left_join(tbl(con, "analysis"), by = c(smpid = "anlsmpidref")) %>%
collect() %>%
select(patpatientid, smpsampleid, smpleukocytes, smpsampledate, smppblymphocytes)
dbDisconnect(con)[1] TRUEplotTab <- pcRes[,1:9] %>% data.frame() %>%
rownames_to_column("patID") %>% as_tibble() %>%
mutate(sampleID = protCLL[,patID]$sampleID) %>%
mutate(perPB = PBtab[match(sampleID, PBtab$smpsampleid),]$smppblymphocytes) %>%
gather(key = "pc", value = "val",-patID,-sampleID,-perPB)
ggplot(plotTab, aes(x=val, y=perPB)) + geom_point() + geom_smooth(method = "lm") +
facet_wrap(~pc, ncol =3, scale = "free")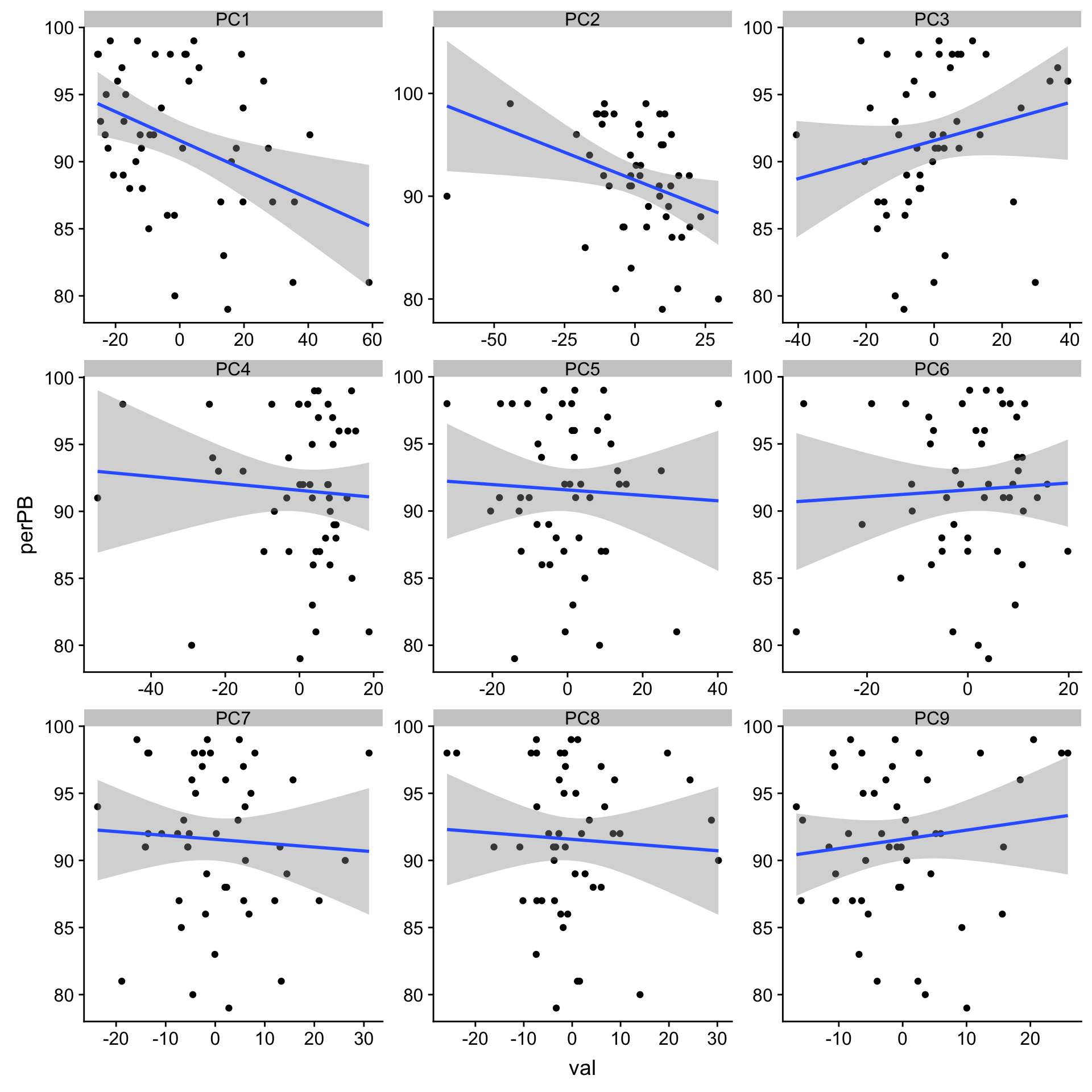
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
corRes <- plotTab %>% group_by(pc) %>% nest() %>%
mutate(m= map(data, ~cor.test(~ val+ perPB,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(pc, estimate, p.value) %>%
arrange(p.value)
corRes# A tibble: 9 x 3
# Groups: pc [9]
pc estimate p.value
<chr> <dbl> <dbl>
1 PC1 -0.398 0.00463
2 PC2 -0.325 0.0228
3 PC3 0.204 0.160
4 PC9 0.126 0.389
5 PC4 -0.0705 0.630
6 PC7 -0.0591 0.687
7 PC8 -0.0557 0.704
8 PC6 0.0527 0.719
9 PC5 -0.0476 0.745 PC1 and PC2 both seem to correlation with %PB. But PC2 is also associate with trisomy12?
Will the correlation with transcriptomics increase if PC1 is regressed out?
pc1 <- pcRes[colnames(rnaMat),1]
corTab <- lapply(geneOverlap, function(n) {
rna <- rnaMat[n,]
pro.q <- proMat.qrilc[n,]
p.single <- anova(lm(rna ~ pro.q))$`Pr(>F)`[1]
p.multi <- car::Anova(lm(rna ~ pro.q + pc1))$`Pr(>F)`[1]
tibble(name = n, corrected = c("no","yes"),
p = c(p.single, p.multi))
}) %>% bind_rows() %>% mutate(p.adj = p.adjust(p, method = "BH")) %>% arrange(p)Number of significant correlations VS FDR cut-off
plotTab <- lapply(seq(0,0.1, length.out = 100), function(fdr) {
filTab <- dplyr::filter(corTab, p.adj < fdr) %>%
group_by(corrected) %>% summarise(n = length(name)) %>% mutate(fdr = fdr)
}) %>% bind_rows()
ggplot(plotTab, aes(x=fdr, y = n, col = corrected))+ geom_line() +
ylab("Number of significant correlations") +
xlab("FDR cut-off")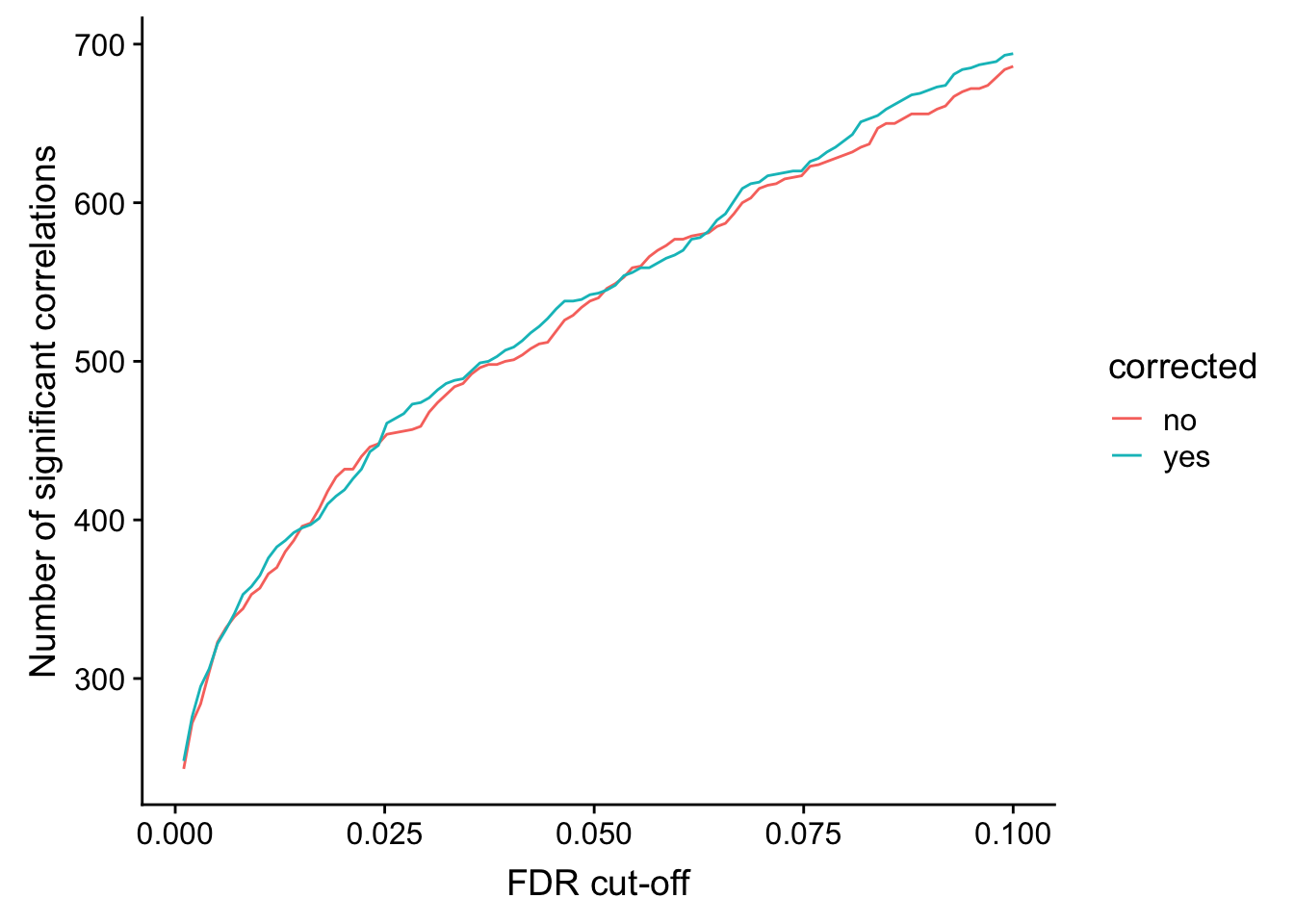
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Seems to improve the correlation a little, but not much. We can include this factor in association test later.
Reconstruct data with PC1 and PC2 removed
protMat <- t(assays(protCLL)[["QRILC"]])
mu <- colMeans(protMat)
Xpca <- prcomp(protMat, center = TRUE, scale. = FALSE)
#reconstruct without the first two components
protMat.new <- Xpca$x[,2:ncol(Xpca$x)] %*% t(Xpca$rotation[,2:ncol(Xpca$x)])
protMat.new <- scale(protMat.new, center = -mu, scale = FALSE)
protMat.new <- t(protMat.new)Hierarchical clustering using reconstructed matrix
plotMat <- protMat.new
colAnno <- colData(protCLL)[,c("gender","IGHV.status","trisomy12")] %>%
data.frame()
plotMat <- jyluMisc::mscale(plotMat, censor = 6)
pheatmap(plotMat, scale = "none", annotation_col = colAnno, clustering_method = "ward.D2",
show_rownames = FALSE, color = colorRampPalette(c("navy","white","firebrick"))(100),
breaks = seq(-6,6, length.out = 101))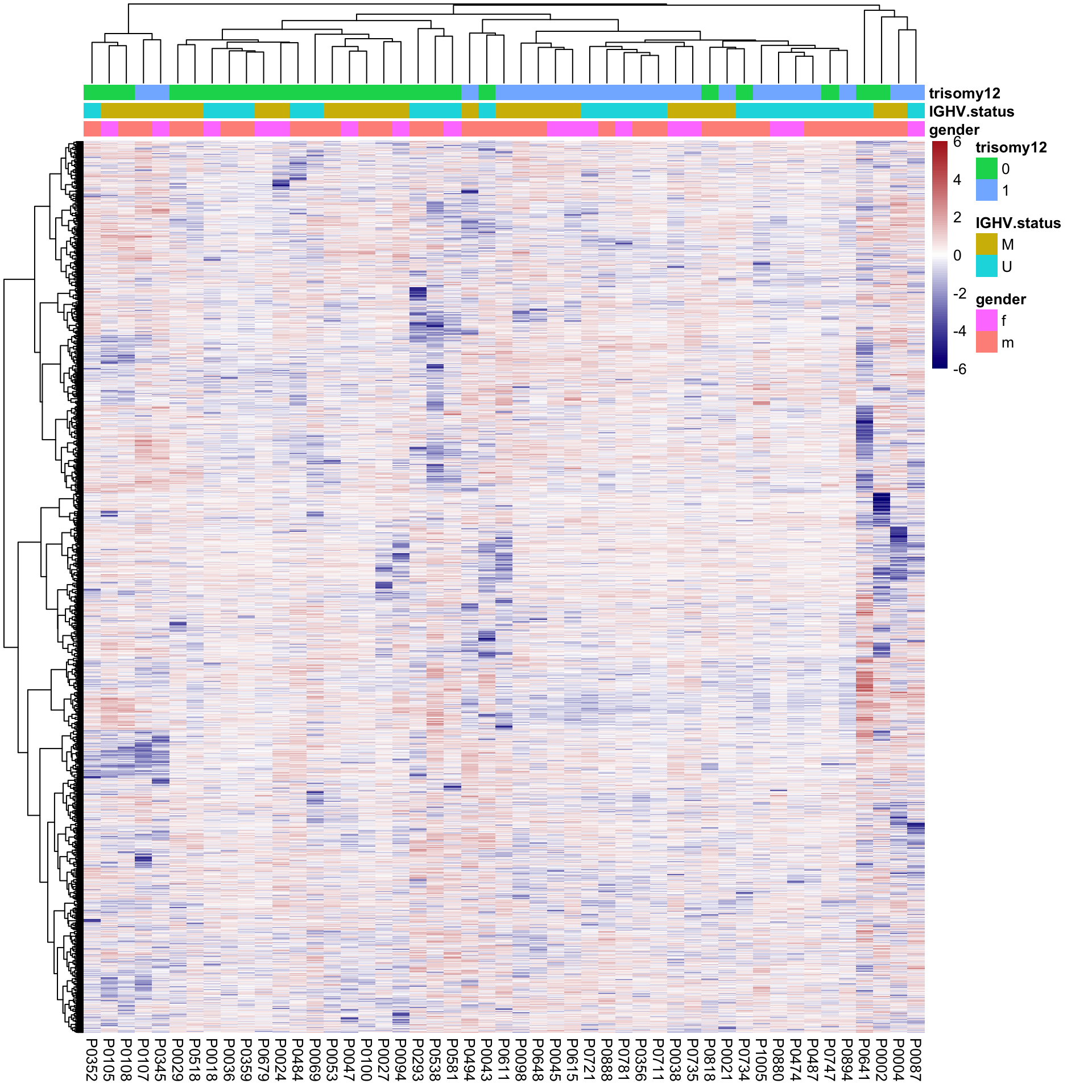
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Better separation for trisomy12 can be observed
PCA using reconstructed matrix
pcOut <- prcomp(t(plotMat), center =TRUE, scale. = FALSE)
pcRes.new <- pcOut$x
eigs <- pcOut$sdev^2
varExp <- structure(eigs/sum(eigs),names = colnames(pcRes.new))
plotTab <- pcRes.new[,1:2] %>% data.frame() %>% cbind(colAnno[rownames(.),]) %>%
rownames_to_column("patID") %>% as_tibble()
ggplot(plotTab, aes(x=PC1, y=PC2, col = IGHV.status, shape = trisomy12)) + geom_point(size=4) +
xlab(sprintf("PC1 (%1.2f%%)",varExp[["PC1"]]*100)) +
ylab(sprintf("PC2 (%1.2f%%)",varExp[["PC2"]]*100))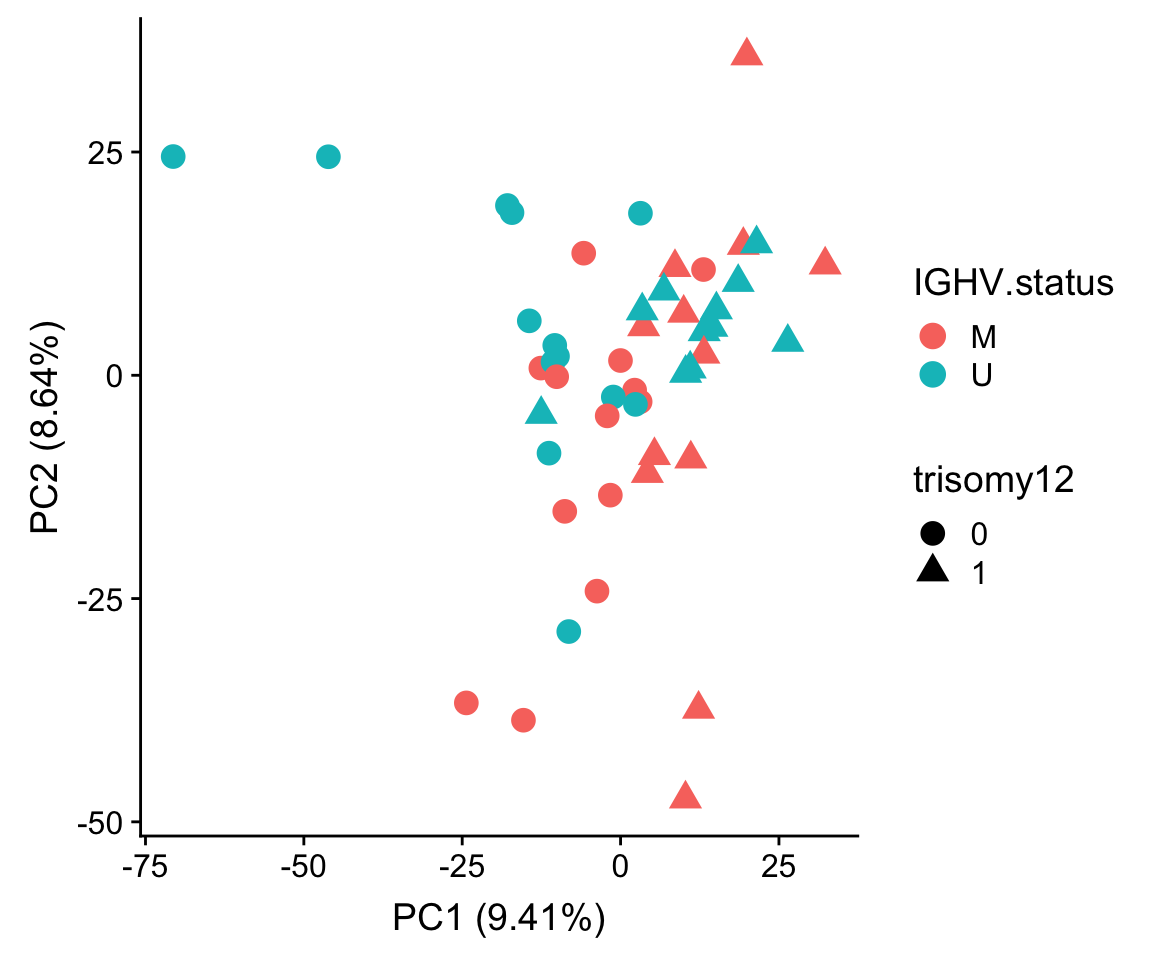
| Version | Author | Date |
|---|---|---|
| 46534c2 | Junyan Lu | 2020-02-27 |
Some outliers dominate the variance
Save the post process object
assays(protCLL)[["QRILC_re"]] <- protMat.new
protCLL$PC1 <- pcRes[colnames(protCLL),1]
protCLL$PC2 <- pcRes[colnames(protCLL),2]
save(protCLL, file = "../output/timsTOF_processed.RData")timsTOF has some quality issues, and it’s not very clear what the major variance in this dataset represents.
sessionInfo()R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DBI_1.0.0 forcats_0.4.0
[3] stringr_1.4.0 dplyr_0.8.3
[5] purrr_0.3.3 readr_1.3.1
[7] tidyr_1.0.0 tibble_2.1.3
[9] tidyverse_1.3.0 DESeq2_1.24.0
[11] SummarizedExperiment_1.14.0 DelayedArray_0.10.0
[13] BiocParallel_1.18.0 matrixStats_0.54.0
[15] Biobase_2.44.0 GenomicRanges_1.36.0
[17] GenomeInfoDb_1.20.0 IRanges_2.18.1
[19] S4Vectors_0.22.0 BiocGenerics_0.30.0
[21] jyluMisc_0.1.5 pheatmap_1.0.12
[23] piano_2.0.2 cowplot_0.9.4
[25] ggplot2_3.2.1 limma_3.40.2
loaded via a namespace (and not attached):
[1] utf8_1.1.4 shinydashboard_0.7.1 tidyselect_0.2.5
[4] RSQLite_2.1.1 AnnotationDbi_1.46.0 htmlwidgets_1.3
[7] grid_3.6.0 maxstat_0.7-25 munsell_0.5.0
[10] codetools_0.2-16 DT_0.7 withr_2.1.2
[13] colorspace_1.4-1 knitr_1.23 rstudioapi_0.10
[16] ggsignif_0.5.0 labeling_0.3 git2r_0.26.1
[19] slam_0.1-45 GenomeInfoDbData_1.2.1 KMsurv_0.1-5
[22] bit64_0.9-7 rprojroot_1.3-2 vctrs_0.2.0
[25] generics_0.0.2 TH.data_1.0-10 xfun_0.8
[28] sets_1.0-18 R6_2.4.0 locfit_1.5-9.1
[31] bitops_1.0-6 fgsea_1.10.0 assertthat_0.2.1
[34] promises_1.0.1 scales_1.0.0 multcomp_1.4-10
[37] nnet_7.3-12 gtable_0.3.0 sandwich_2.5-1
[40] workflowr_1.6.0 rlang_0.4.1 zeallot_0.1.0
[43] genefilter_1.66.0 cmprsk_2.2-8 splines_3.6.0
[46] lazyeval_0.2.2 acepack_1.4.1 broom_0.5.2
[49] checkmate_1.9.3 yaml_2.2.0 abind_1.4-5
[52] modelr_0.1.5 crosstalk_1.0.0 backports_1.1.4
[55] httpuv_1.5.1 Hmisc_4.2-0 tools_3.6.0
[58] relations_0.6-8 RPostgreSQL_0.6-2 ellipsis_0.2.0
[61] gplots_3.0.1.1 RColorBrewer_1.1-2 Rcpp_1.0.1
[64] base64enc_0.1-3 visNetwork_2.0.7 zlibbioc_1.30.0
[67] RCurl_1.95-4.12 ggpubr_0.2.1 rpart_4.1-15
[70] zoo_1.8-6 ggrepel_0.8.1 haven_2.2.0
[73] cluster_2.1.0 exactRankTests_0.8-30 fs_1.3.1
[76] magrittr_1.5 data.table_1.12.2 openxlsx_4.1.0.1
[79] reprex_0.3.0 survminer_0.4.4 mvtnorm_1.0-11
[82] whisker_0.3-2 hms_0.5.2 shinyjs_1.0
[85] mime_0.7 evaluate_0.14 xtable_1.8-4
[88] XML_3.98-1.20 rio_0.5.16 readxl_1.3.1
[91] gridExtra_2.3 compiler_3.6.0 KernSmooth_2.23-15
[94] crayon_1.3.4 htmltools_0.3.6 later_0.8.0
[97] Formula_1.2-3 geneplotter_1.62.0 lubridate_1.7.4
[100] dbplyr_1.4.2 MASS_7.3-51.4 Matrix_1.2-17
[103] car_3.0-3 cli_1.1.0 marray_1.62.0
[106] gdata_2.18.0 igraph_1.2.4.1 pkgconfig_2.0.2
[109] km.ci_0.5-2 foreign_0.8-71 xml2_1.2.2
[112] annotate_1.62.0 XVector_0.24.0 drc_3.0-1
[115] rvest_0.3.5 digest_0.6.19 rmarkdown_1.13
[118] cellranger_1.1.0 fastmatch_1.1-0 survMisc_0.5.5
[121] htmlTable_1.13.1 curl_3.3 shiny_1.3.2
[124] gtools_3.8.1 lifecycle_0.1.0 nlme_3.1-140
[127] jsonlite_1.6 carData_3.0-2 fansi_0.4.0
[130] pillar_1.4.2 lattice_0.20-38 httr_1.4.1
[133] plotrix_3.7-6 survival_2.44-1.1 glue_1.3.1
[136] zip_2.0.2 bit_1.1-14 stringi_1.4.3
[139] blob_1.1.1 latticeExtra_0.6-28 caTools_1.17.1.2
[142] memoise_1.1.0