Process each omics data of T cells
Junyan Lu
Last updated: 2024-04-08
Checks: 5 1
Knit directory: RA_Tcell_omics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load libraries
library(vsn)
library(jyluMisc)
library(SummarizedExperiment)
library(tidyverse)
knitr::opts_chunk$set(warning = FALSE, message = FALSE)Load sample annotations
patTab1 <- readxl::read_xlsx("../data/Data_2023-02-16/MultiOmics Patien Data.xlsx", sheet = 1) %>%
filter(!is.na(Pseudonyme)) %>%
dplyr::rename(sampleID = Pseudonyme) %>%
mutate(sampleID = str_remove_all(sampleID," ")) %>%
mutate(group = "RA") %>%
dplyr::rename(`Date of Birth` = `Date of Birtrh`)
patTab2 <- readxl::read_xlsx("../data/Data_2023-02-16/MultiOmics Patien Data.xlsx", sheet = 2) %>%
filter(!is.na(Pseudonyme)) %>%
dplyr::rename(sampleID = Pseudonyme, `Date of sample` = `Date of Sample`) %>%
mutate(sampleID = str_remove_all(sampleID," ")) %>%
mutate(group = "HC") %>%
mutate(`Date of Birth` = as.Date(as.character(`Date of Birth`), format = "%Y"))
patTab <- bind_rows(patTab1, patTab2) %>%
distinct(sampleID, .keep_all = TRUE)Choose useful data
patTab <- mutate(patTab, Age = as.numeric((`Date of sample` - `Date of Birth`)/365),
dateMeta = as.character(Metabolites),
dateProt = as.character(Proteome),
datePhos = as.character(Phosphoproteome),
dateMeth = as.character(Methylation),
dateFACS = as.character(Flowcytometry)) %>%
dplyr::rename(BMI = `BMI(kg/(m^2))`, CRP = `CRP [mg/l]`) %>%
mutate(CRP = as.numeric(str_remove_all(CRP, "<"))) %>%
select(sampleID, Gender, Age, CCP, RF, GC, MTX, Leflunomid, Sulfasalazin, Quensyl, BMI, CRP,DAS28, group,
dateMeta, dateProt, datePhos, dateMeth, dateFACS)Sample metadata table
patTab %>% mutate_if(is.numeric, formatC, digits=1) %>% DT::datatable()Process the the new metabolic table
metaTab1 <- readxl::read_xlsx("../data/Data_2023-02-16/Metabolites/20211022_results_GCMS_metabolomics_CD8_RAHC.xlsx", sheet = 1, skip = 1) %>%
dplyr::rename(sampleID = `...1`) %>%
pivot_longer(-sampleID, names_to = "feature", values_to = "count") %>%
mutate(sheet = "Metabolites")
metaTab2 <- readxl::read_xlsx("../data/Data_2023-02-16/Metabolites/20211022_results_GCMS_metabolomics_CD8_RAHC.xlsx", sheet = 2, skip = 1) %>%
dplyr::rename(sampleID = ProbenID) %>%
pivot_longer(-sampleID, names_to = "feature", values_to = "count") %>%
mutate(sheet = "AA-AC")
metaTab <- bind_rows(metaTab1, metaTab2) %>%
filter(!is.na(sampleID)) %>%
left_join(patTab, by = "sampleID") %>%
mutate(metabolite = feature)
seMeta <- jyluMisc::tidyToSum(metaTab, rowID = "feature", colID = "sampleID", values = "count", annoRow = c("sheet","metabolite"),
annoCol = colnames(patTab)[2:ncol(patTab)])
dim(seMeta)[1] 65 36Why there are two sheets in the excel table, are there any difference in the measurement process? What is the difference between this data and previous data? Seems just more samples, the values are the same. Are they from different batches?
QC
plotTab <- metaTab
metaMat <- assay(seMeta )Feature distribution
Per-sample
Raw scale
ggplot(plotTab, aes(x=sampleID, y=count, fill = group)) +
geom_boxplot() + geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))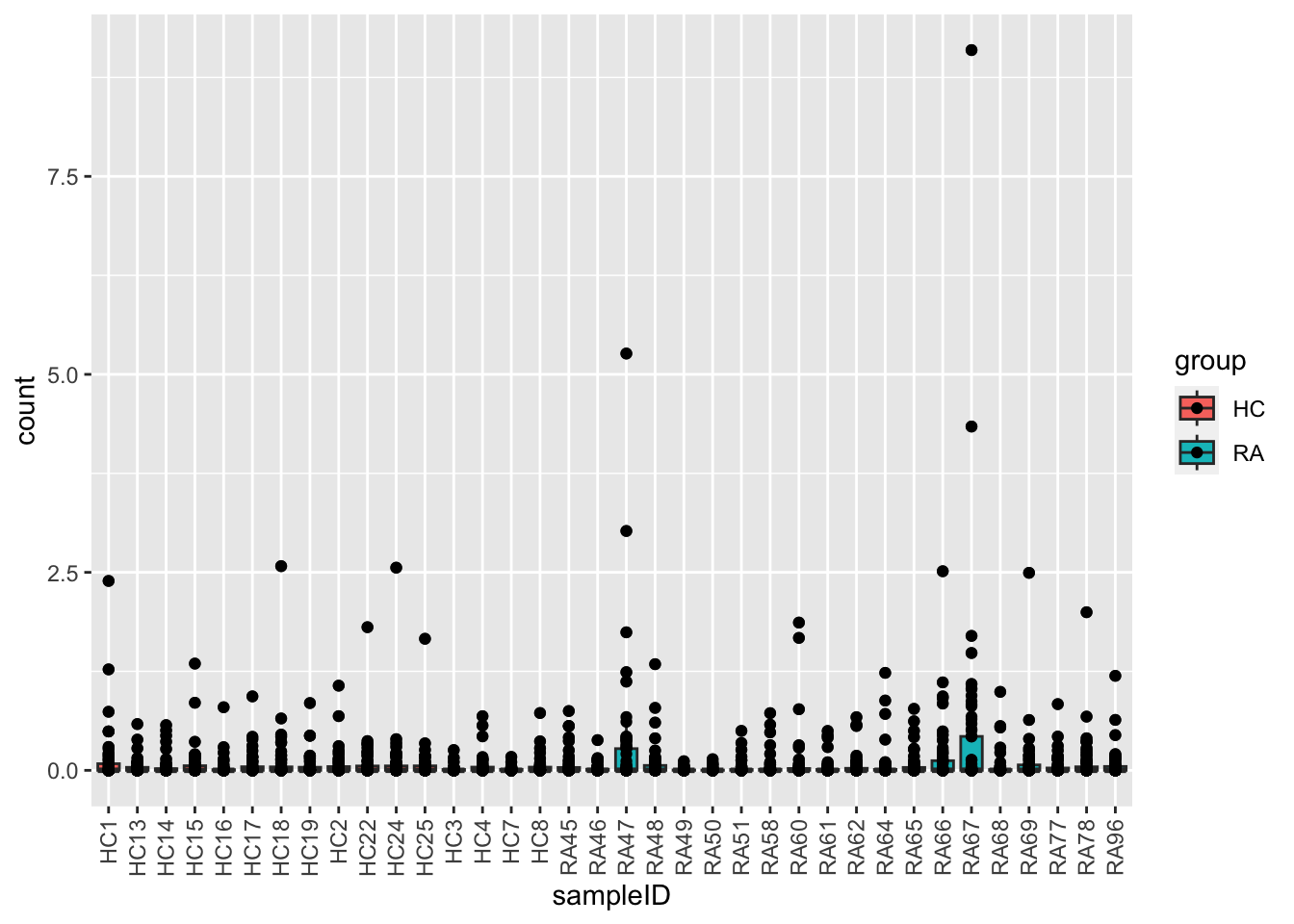
glog transformed
ggplot(plotTab, aes(x=sampleID, y=glog(count), fill = group)) +
geom_boxplot() + geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))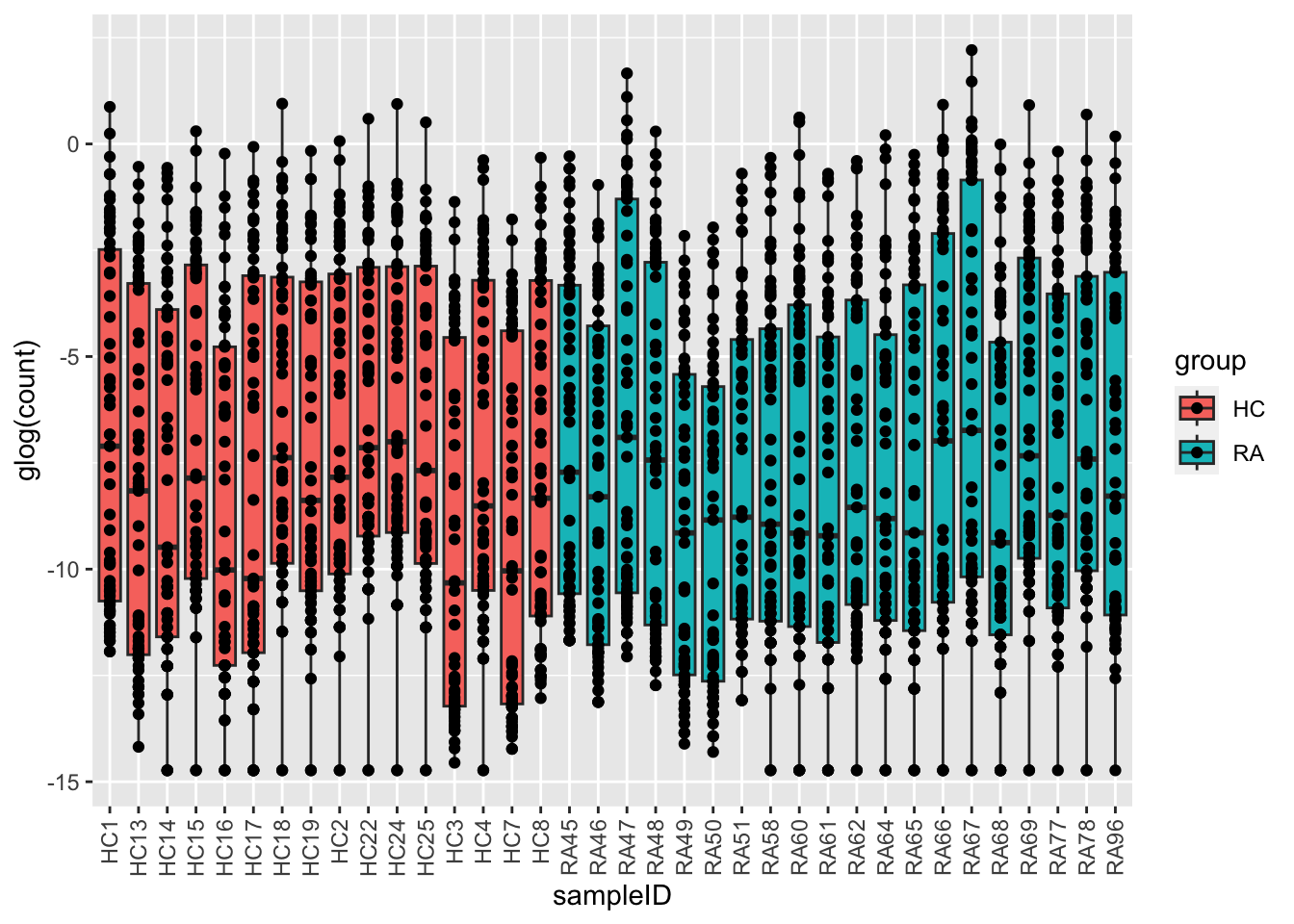
Per feature
Raw scale
ggplot(plotTab, aes(x=feature, y=count)) +
geom_boxplot() + geom_point(aes(col=group)) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))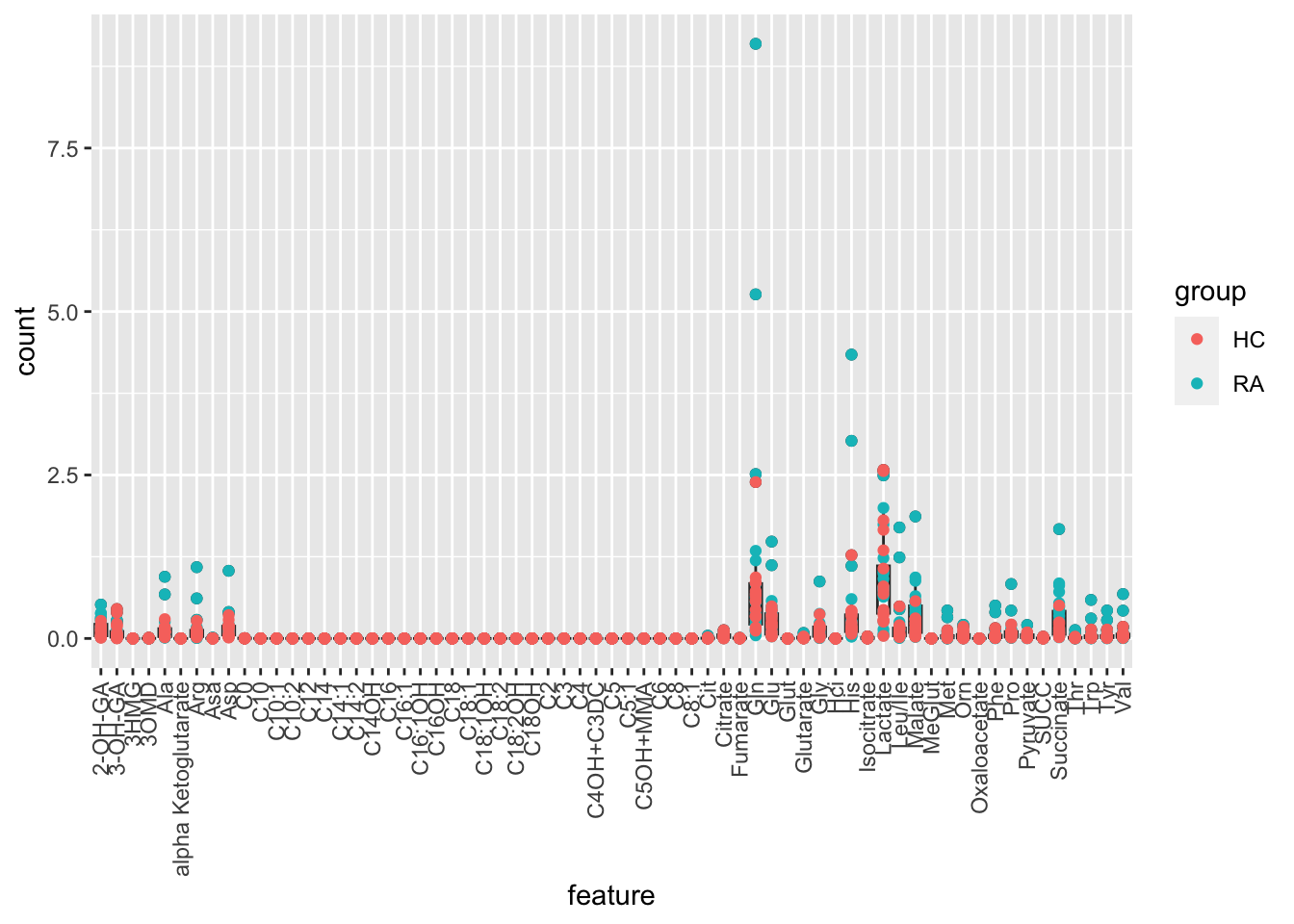
glog transformed
ggplot(plotTab, aes(x=feature, y=glog(count))) +
geom_boxplot() + geom_point(aes(col= group)) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))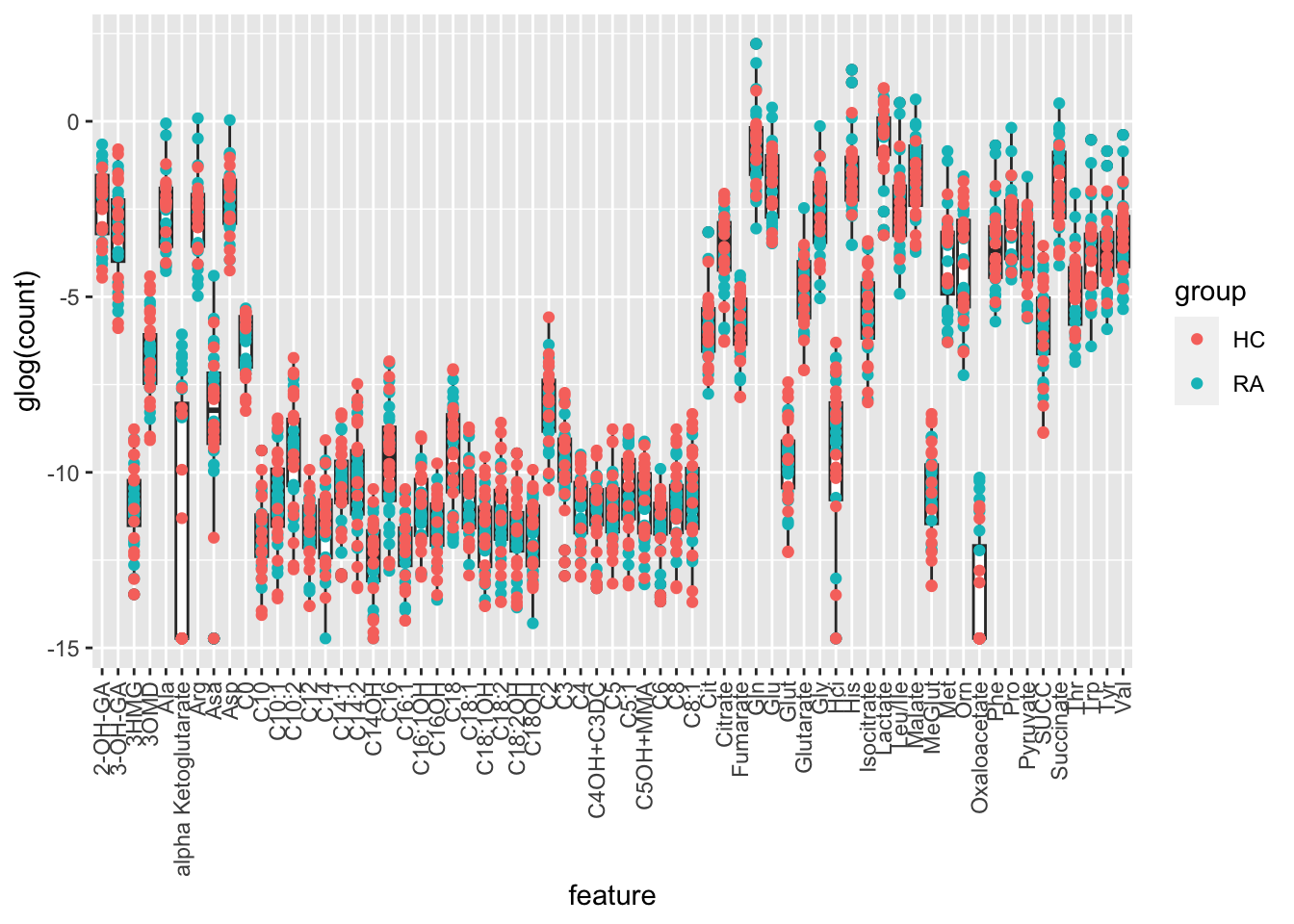
PCA
Raw scale
metaMat <- metaMat[,complete.cases(t(metaMat))]
pcRes <- prcomp(t(metaMat), scale. = FALSE, center = TRUE)
plotTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patTab, by = "sampleID")
ggplot(plotTab, aes(x=PC1, y=PC2, col = group, label = sampleID)) +
geom_point() +
geom_text()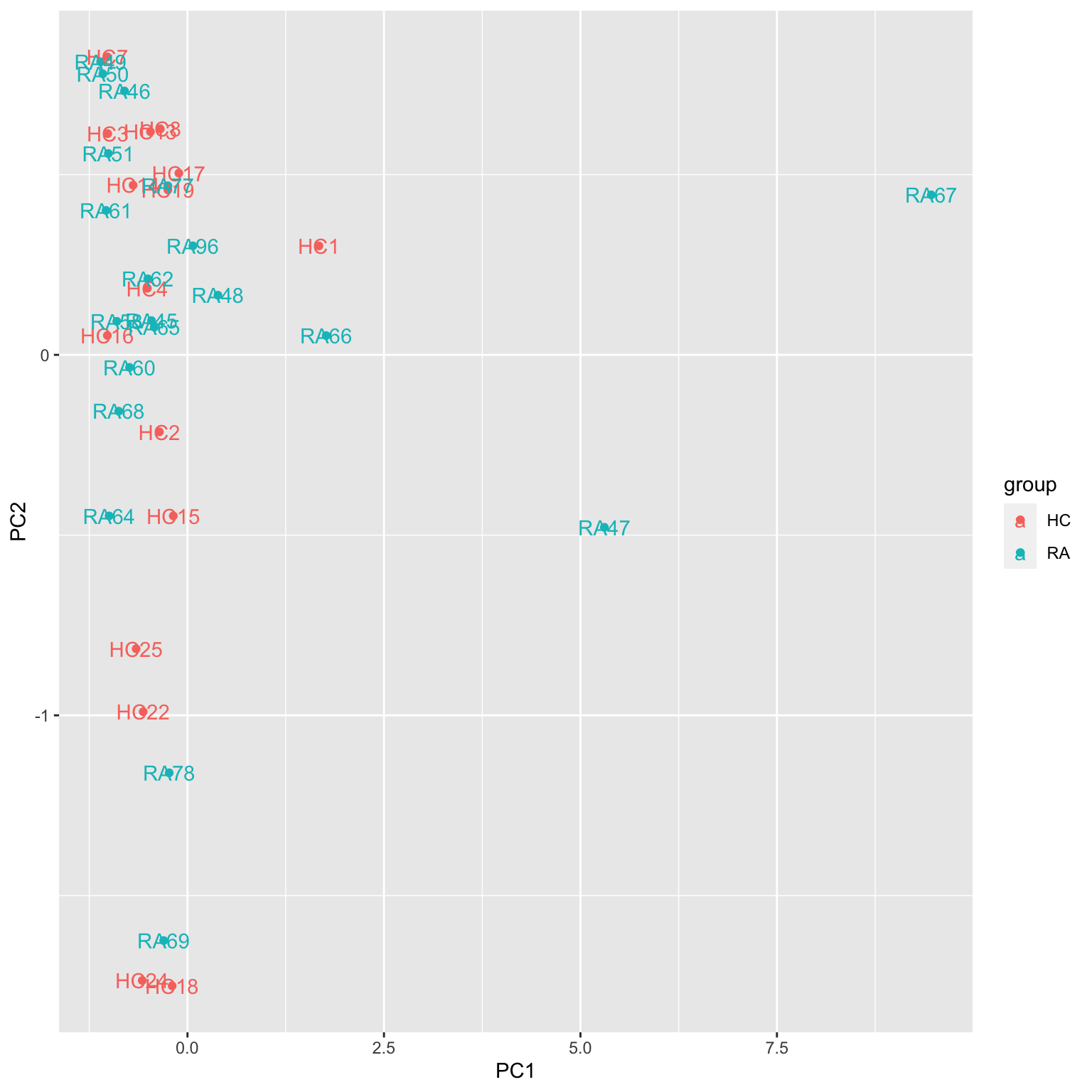
glog transformed
Colored by phenotype
metaMat <- jyluMisc::glog(metaMat)
pcRes <- prcomp(t(metaMat), scale. = FALSE, center = TRUE)
plotTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patTab, by = "sampleID")
ggplot(plotTab, aes(x=PC1, y=PC2, col = group, label = sampleID)) +
geom_point() +
geom_text()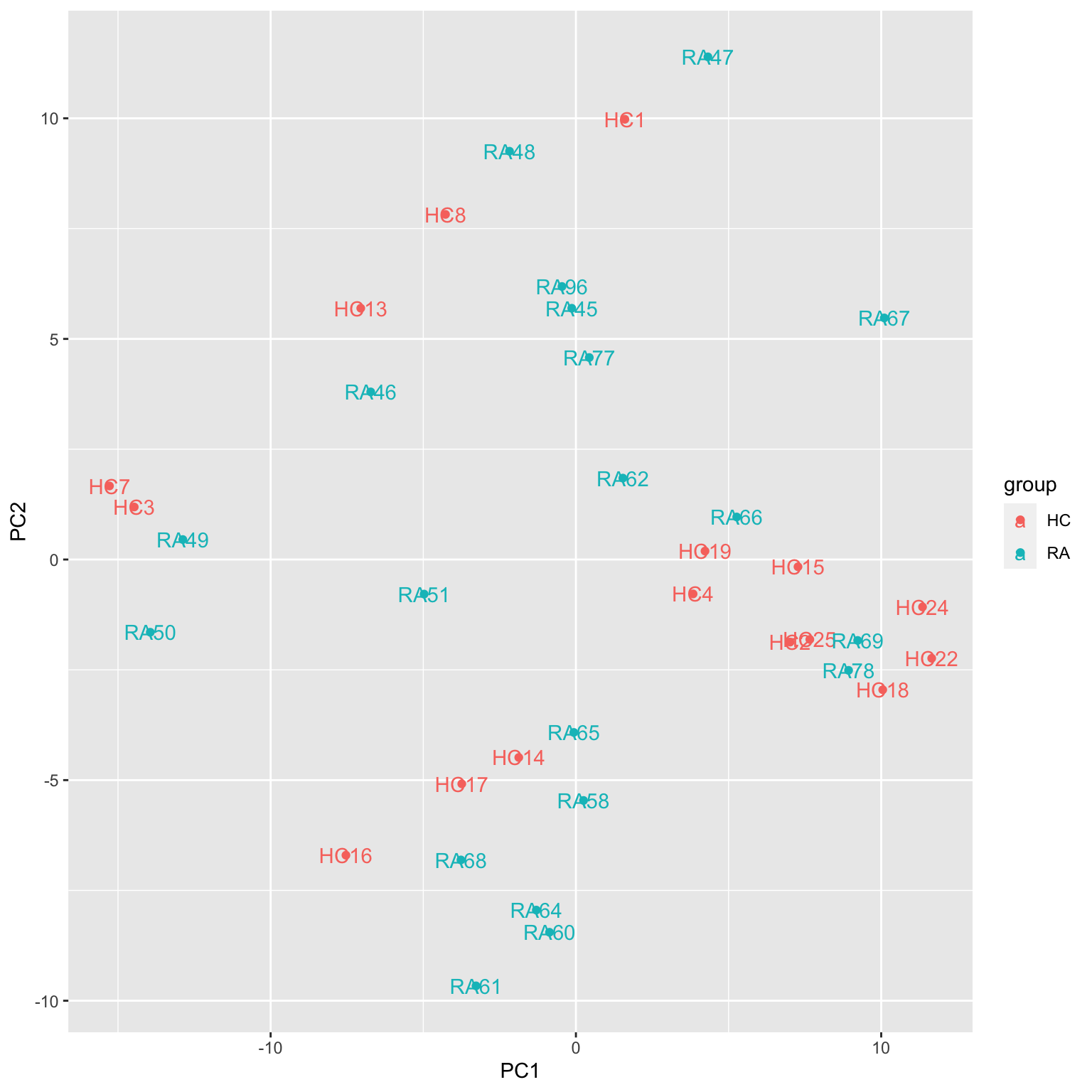
Colored by date of metabolic measurement
metaMat <- jyluMisc::glog(metaMat)
pcRes <- prcomp(t(metaMat), scale. = FALSE, center = TRUE)
plotTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patTab, by = "sampleID")
ggplot(plotTab, aes(x=PC1, y=PC2, col = dateMeta, label = sampleID)) +
geom_point() +
geom_text()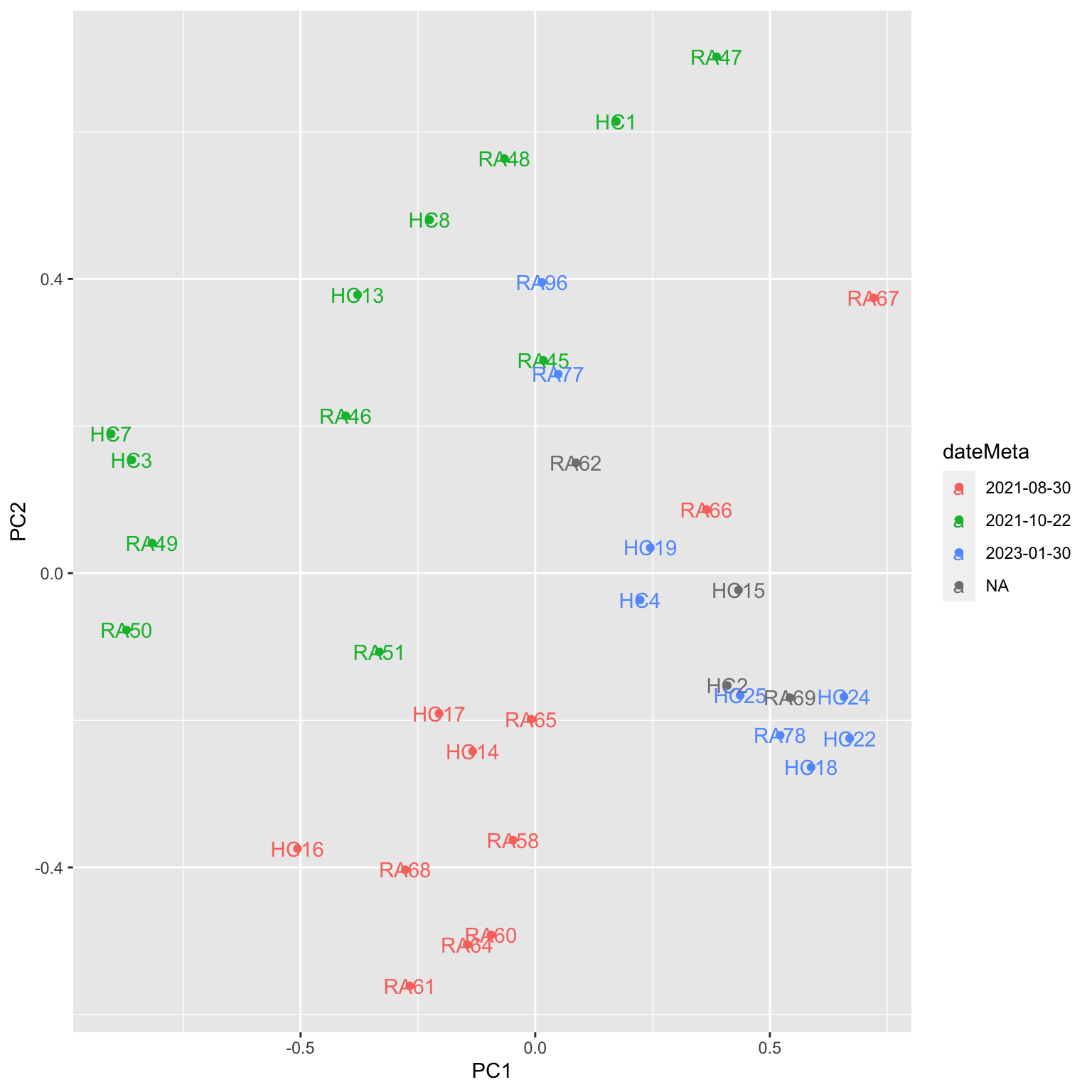 Batch effect could be observed
Batch effect could be observed
Two samples don’t have date information
Process Proteomics data
Map the sample name
sampleMap <- readxl::read_xlsx("../data/Data_2023-02-16/Proteome_Phosphome/sample submission naming.xlsx", col_names = c("id","sampleID")) %>%
mutate(id = sprintf("%s%02d","Sample",id))Read in additional proteomic sample metadata
smpMeta <- readxl::read_xlsx("../data/TF0489/Sample-Information.xlsx") %>%
filter(!is.na(`Patient Group`))
smpMeta <- smpMeta[,c(1,5,6,7,8)]
colnames(smpMeta) <- c("sampleID","protConc","quantStart","sampleVol","bufferComp")
smpMeta <- mutate(smpMeta, sampleID = paste0("Sample",sprintf("%02s",sampleID)))It seems compared to previous proteomic data, in this data, the replicates are removed and buffer A was prioritized.
Read data table
protTab <- readxl::read_xlsx("../data/Data_2023-02-16/Proteome_Phosphome/TF0489-3_results/TF0489-3_filtered_proteinGroups.xlsx") %>%
filter(!`Potential contaminant` %in% "+") %>%
select(`Majority protein IDs`, `Gene names`, contains("LFQ intensity")) %>%
dplyr::rename(name = "Majority protein IDs", symbol = "Gene names") %>%
pivot_longer(-c(name, symbol), names_to = "sampleID", values_to = "count") %>%
mutate(sampleID = str_remove(sampleID, "LFQ intensity ")) %>%
filter(count>0) %>% left_join(smpMeta, by = "sampleID") %>%
mutate(sampleID = sampleMap[match(sampleID,sampleMap$id),]$sampleID) %>%
left_join(patTab, by = "sampleID")
idMap <- unique(protTab$name)
idMap <- structure(paste0("prot",seq_along(idMap)), names = idMap)
protTab <- mutate(protTab, ID = idMap[name])Choose the first symbol if multiple symbols are present in the symbol column
# Get the last symbol of a protein that has multiple gene symbols
getOneSymbol <- function(Gene) {
outStr <- sapply(Gene, function(x) {
sp <- str_split(x, ";")[[1]]
sp[length(sp)]
})
names(outStr) <- NULL
outStr
}
protTab$symbol <- getOneSymbol(protTab$symbol)
#only keep proteins with symbols
protTab <- filter(protTab, !symbol %in% c("",NA))Created summarised experiment
seProt <- jyluMisc::tidyToSum(protTab, "ID", "sampleID","count",
annoRow = c("ID","name","symbol"),
annoCol = c(colnames(patTab),"protConc", "quantStart", "sampleVol", "bufferComp"))QA
Data completeness per sample
countMat <- assay(seProt)
plotTab <- tibble(sample = colnames(seProt),
perNA = colSums(is.na(countMat))/nrow(countMat))
ggplot(plotTab, aes(x=sample, y=1-perNA)) +
geom_bar(stat = "identity") +
ylab("completeness") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust=0))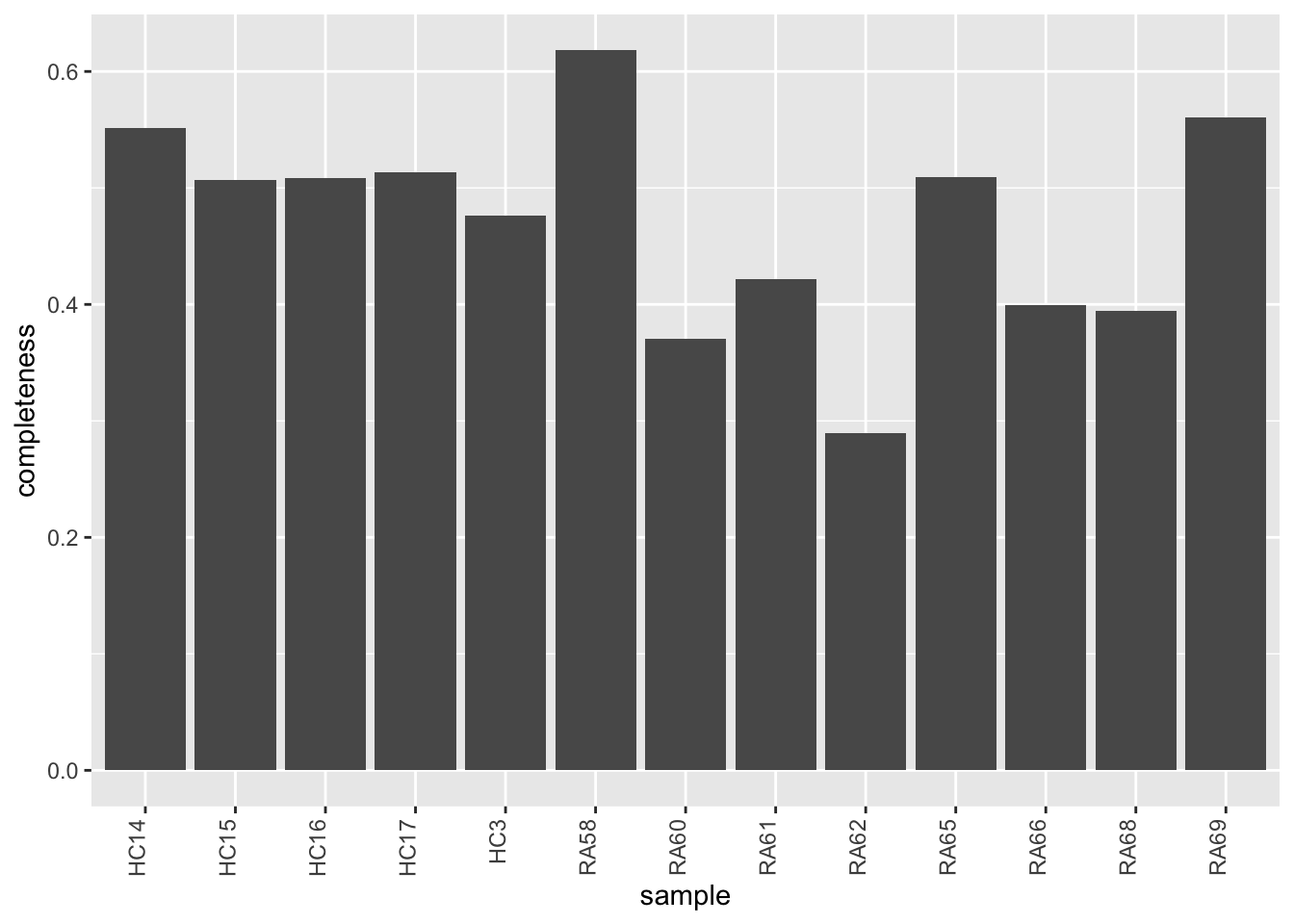
Plot a cumulative curve of missing value cut-off and remaining number of features
missRate <- tibble(id = rownames(countMat),
rate = rowSums(is.na(countMat))/ncol(countMat))
cumTab <- lapply(seq(0,1,0.05), function(cutRate) {
tibble(cut= cutRate,
per = sum(missRate$rate <= cutRate)/nrow(missRate))
} ) %>%
bind_rows()
ggplot(cumTab, aes(x=cut,y=per)) +
geom_line() +
xlab("Allowed missing value rate") +
ylab("Percentage of remaining features")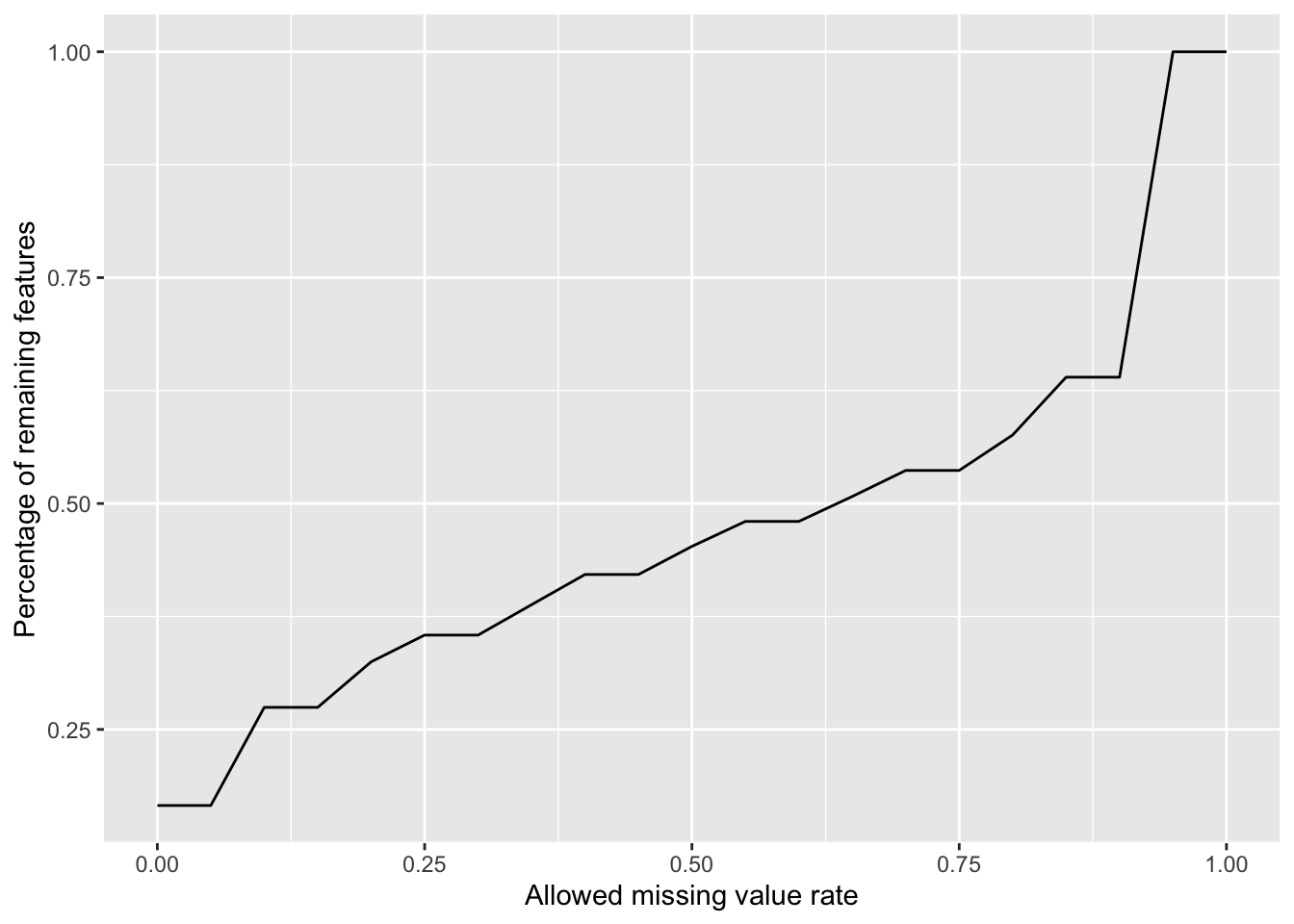 Missing value heatmap to check missing value structure
Missing value heatmap to check missing value structure
Visualize the missing value pattern
DEP::plot_missval(seProt) Looks pretty sparse, maybe due to the DDA data aquisition method
Looks pretty sparse, maybe due to the DDA data aquisition method
Keep proteins detected in at least half of the sample (missing rate <= 0.5)
protFilt <- seProt[filter(missRate, rate <=0.5)$id,]
dim(protFilt)[1] 2342 13Data distribution
countMat <- assay(protFilt)
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value)) %>%
mutate(log2Val = log2(value))ggplot(countTab, aes(x=name, y=log2Val)) +
geom_boxplot() + geom_point()
Imputation and normalization
Vst
protMat <- assay(protFilt)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
protNorm <- protFilt
assay(protNorm) <- normMatImputation
protImp <- DEP::impute(protNorm, "QRILC")
assays(protFilt)[["norm"]] <- normMat
assays(protFilt)[["imputed"]] <- assay(protImp)
rowData(protFilt) <- rowData(protImp)Distribution after normalizaiton
countMat <- assays(protFilt)[["imputed"]]
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value))
ggplot(countTab, aes(x=name, y=value)) +
geom_boxplot() + geom_point()
Mean versus variant plot
plotTab <- tibble(meanVal = rowMeans(countMat),
var = apply(countMat, 1, var))
ggplot(plotTab, aes(x=meanVal,y=var)) +
geom_point()
Heatmap visualization
library(pheatmap)
#select top 1000 most variant
colAnno <- colData(protFilt) %>% data.frame()
colAnno <- colAnno[,c("Gender","group","dateProt","protConc","quantStart","sampleVol","bufferComp")]
#colAnno[["sampleName"]] <- NULL
plotMat <- countMat[order(plotTab$var, decreasing = TRUE)[1:1000],]
pheatmap(plotMat, show_rownames = FALSE, scale = "row",
annotation_col = colAnno,
clustering_method = "ward.D2")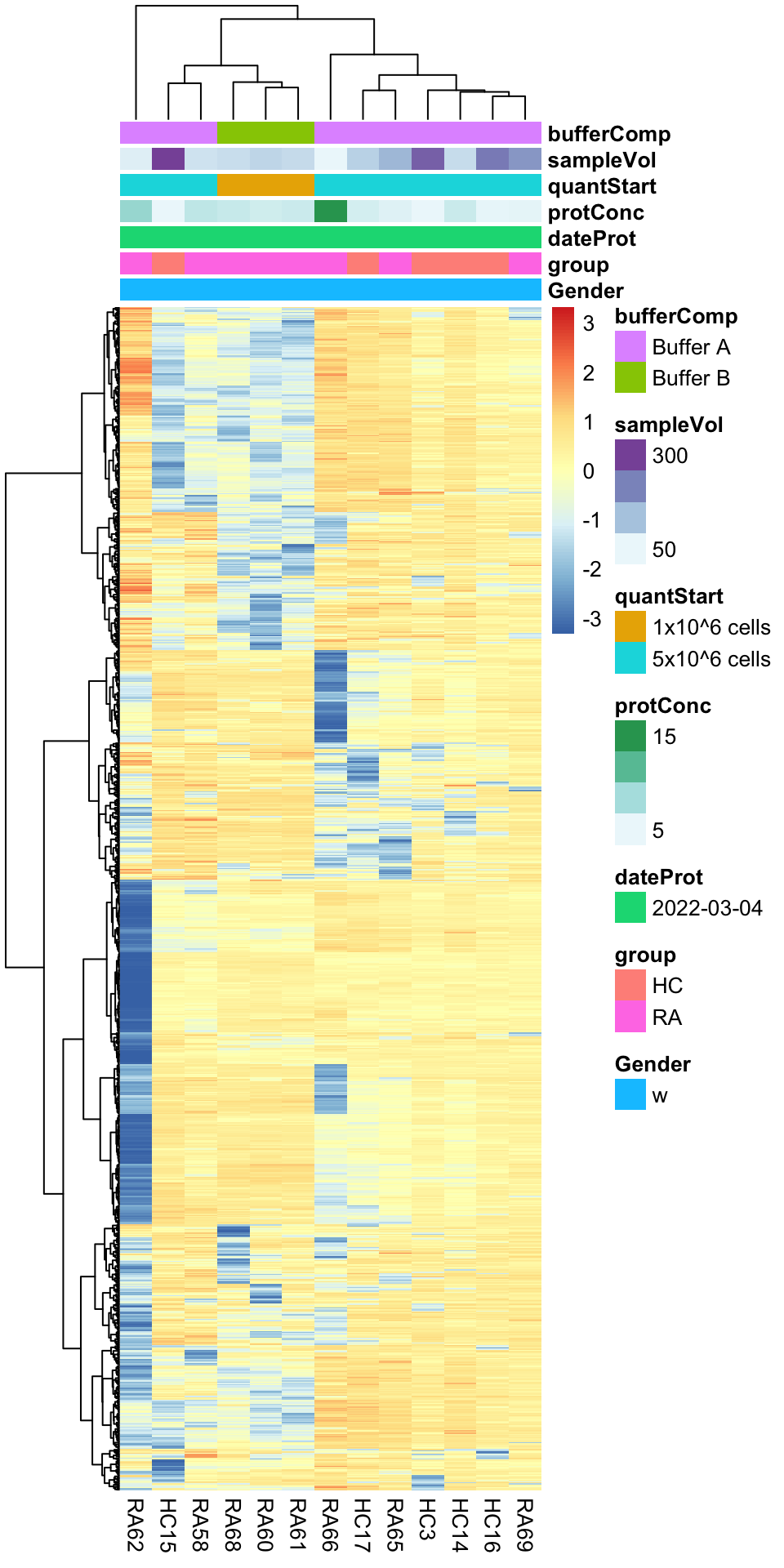 Buffer composition and startQuant could be potential counfunding
factor
Buffer composition and startQuant could be potential counfunding
factor
PCA
prRes <- prcomp(t(plotMat), scale. = FALSE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))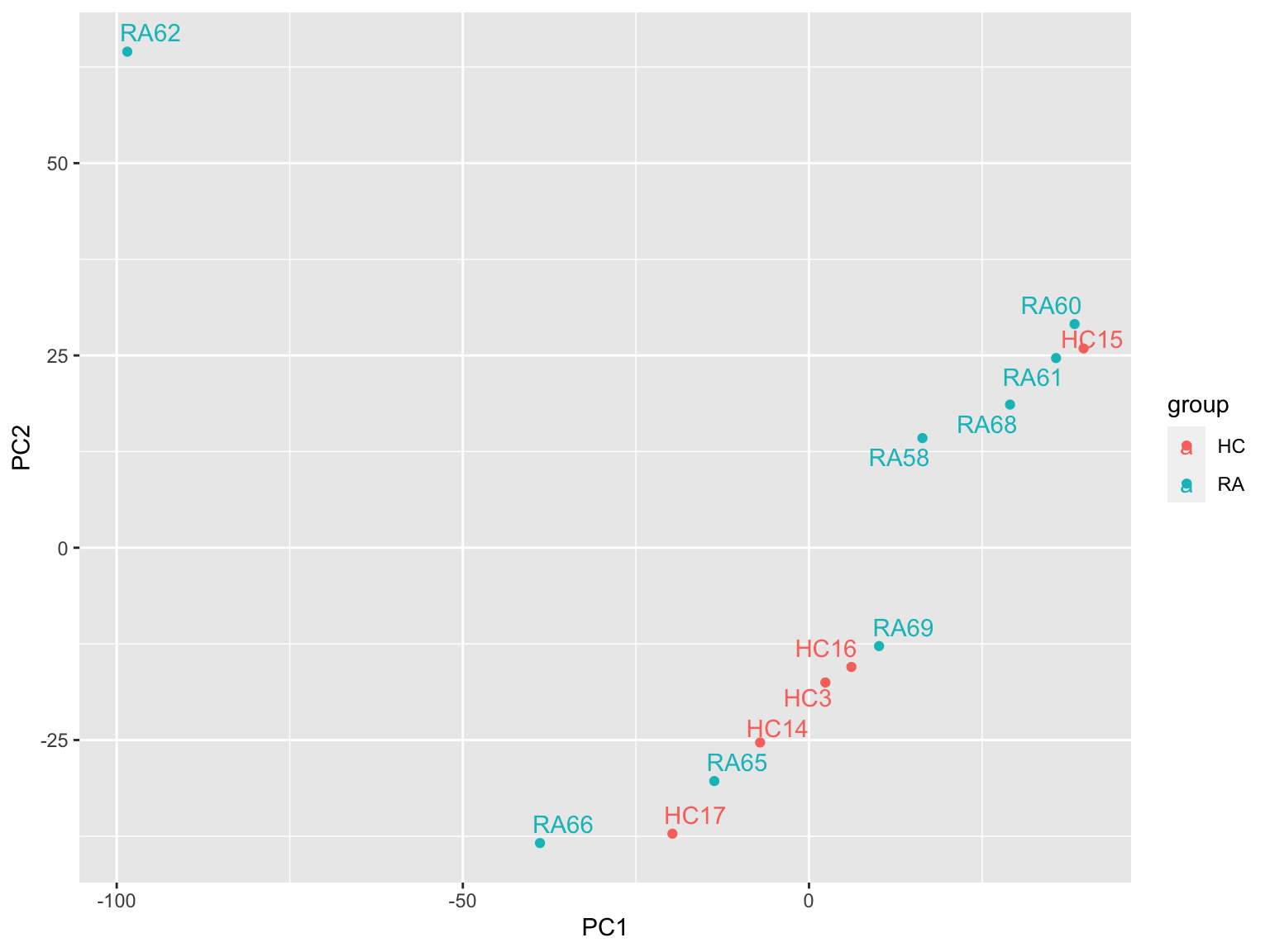 RA62 looks like an outlier
RA62 looks like an outlier
Remove one potential outlier, RA62 and redo the preprocessing
Subset
protSub <- seProt[,seProt$sampleID != "RA62"]
#remove proteins with more than 50% missing values
protSub <- protSub[rowSums(is.na(assay(protSub)))/ncol(protSub)<=0.5,]Vst
protMat <- assay(protSub)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
protNorm <- protSub
assay(protNorm) <- normMatImputation
protImp <- DEP::impute(protNorm, "QRILC")
assays(protSub)[["norm"]] <- normMat
assays(protSub)[["imputed"]] <- assay(protImp)Redo pca
Color by group
plotMat <- assays(protSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))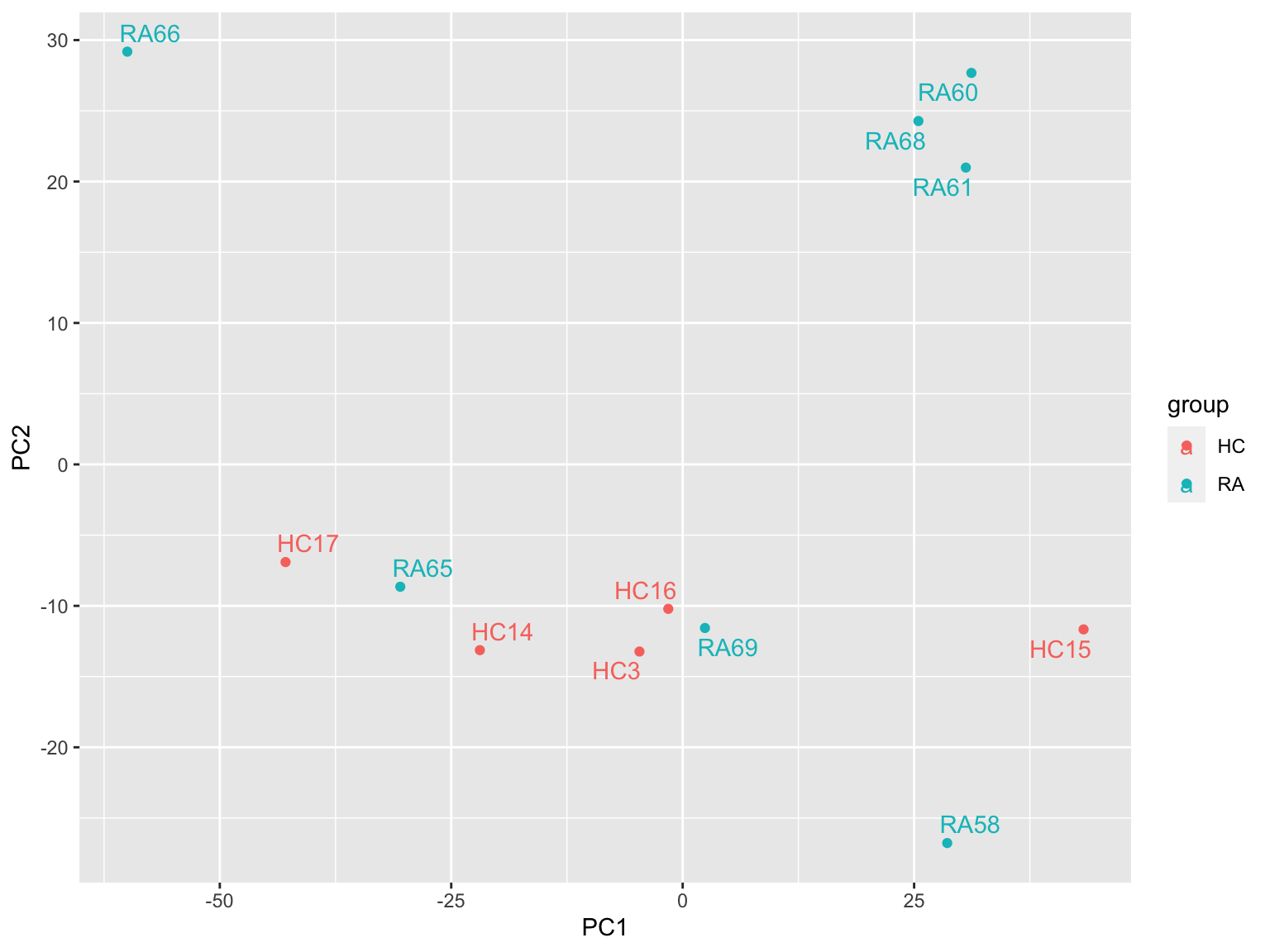 Looks better.
Looks better.
Color by buffer composition
plotMat <- assays(protSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = bufferComp)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))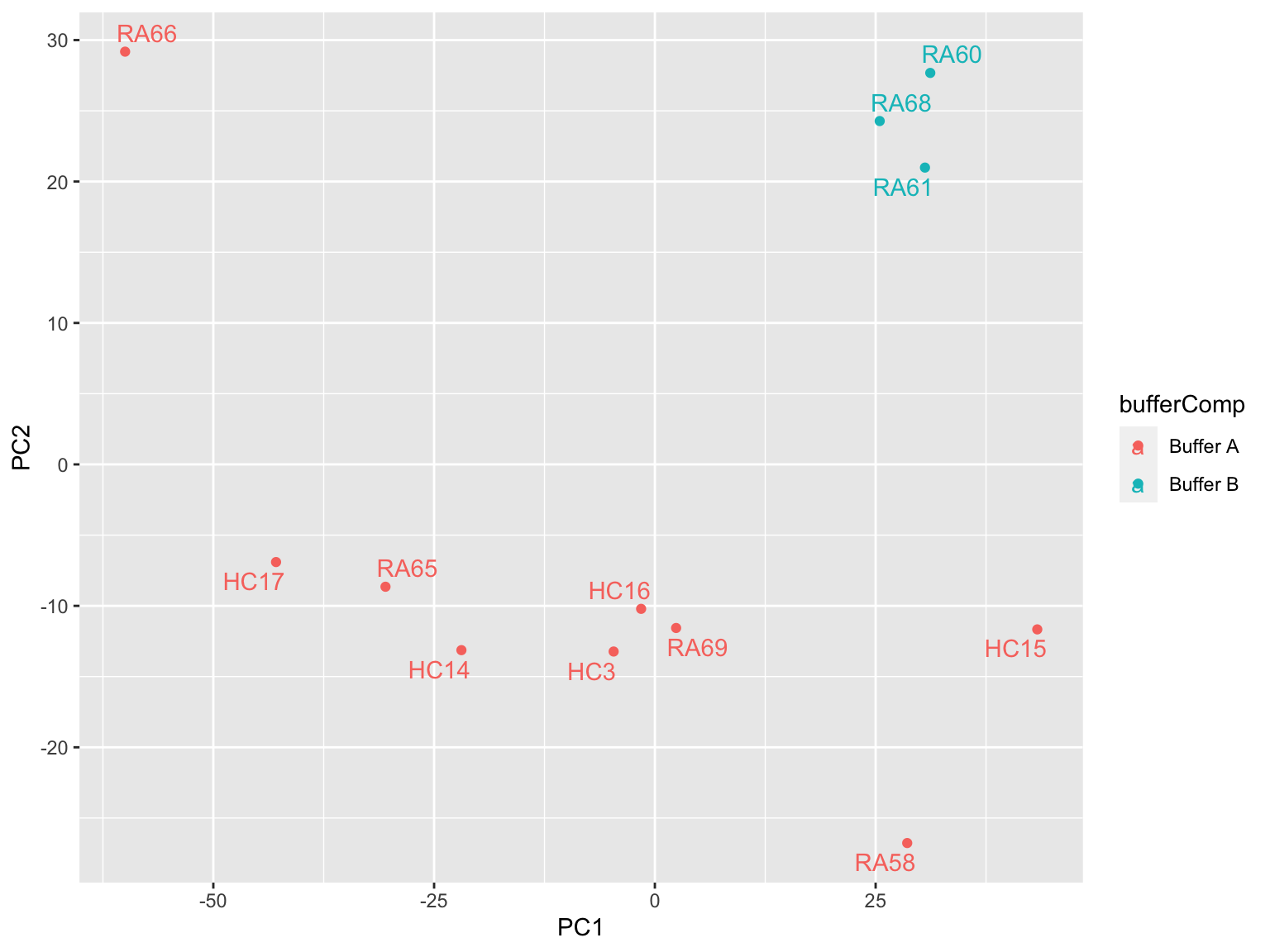
Phospho-proteomics
Read data table
phosTab <- readxl::read_xlsx("../data/Data_2023-02-16/Proteome_Phosphome/TF0489-3_results/TF0489-3_filtered_PhosphoSTY.xlsx") %>%
filter(!`Potential contaminant` %in% "+") %>%
select(Proteins, `Leading proteins`, Position, `Gene names`, `Amino acid`,
matches("(Intensity|Score|Score diff|Localization prob) Sample..$")) %>%
pivot_longer(matches("Sample..$")) %>%
mutate(sampleID = str_extract(name, "Sample.."),
type = str_remove(name," Sample..")) %>%
select(-name) %>%
pivot_wider(names_from = type, values_from = value) %>%
filter(`Localization prob` >= 0.75,
`Score diff` >= 5,
Score >= 10,
Intensity >0) %>%
select(-c(`Localization prob`, `Score diff`, Score)) %>%
left_join(smpMeta, by = "sampleID") %>%
mutate(sampleID = sampleMap[match(sampleID,sampleMap$id),]$sampleID) %>%
dplyr::rename(name = "Leading proteins", symbol = "Gene names", AA = "Amino acid", count = "Intensity") %>%
mutate(symbol = getOneSymbol(symbol)) %>%
mutate(siteID = paste0(Proteins,"_",Position),
site = paste0(symbol,"_",AA,Position)) %>%
filter(!symbol %in% c(NA,""))
idMap <- unique(phosTab$siteID)
idMap <- structure(paste0("phos",seq_along(idMap)), names = idMap)
phosTab <- mutate(phosTab, ID = idMap[siteID]) %>%
left_join(patTab, by = "sampleID") Created summarised experiment
sePhos <- jyluMisc::tidyToSum(phosTab, "ID", "sampleID","count",
annoRow = c("ID","name","symbol", "site","siteID", "Position", "AA"),
annoCol = c(colnames(patTab),"protConc", "quantStart", "sampleVol", "bufferComp"))QA
Data completeness per sample
countMat <- assay(sePhos)
plotTab <- tibble(sample = colnames(sePhos),
perNA = colSums(is.na(countMat))/nrow(countMat))
ggplot(plotTab, aes(x=sample, y=1-perNA)) +
geom_bar(stat = "identity") +
ylab("completeness") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust=0))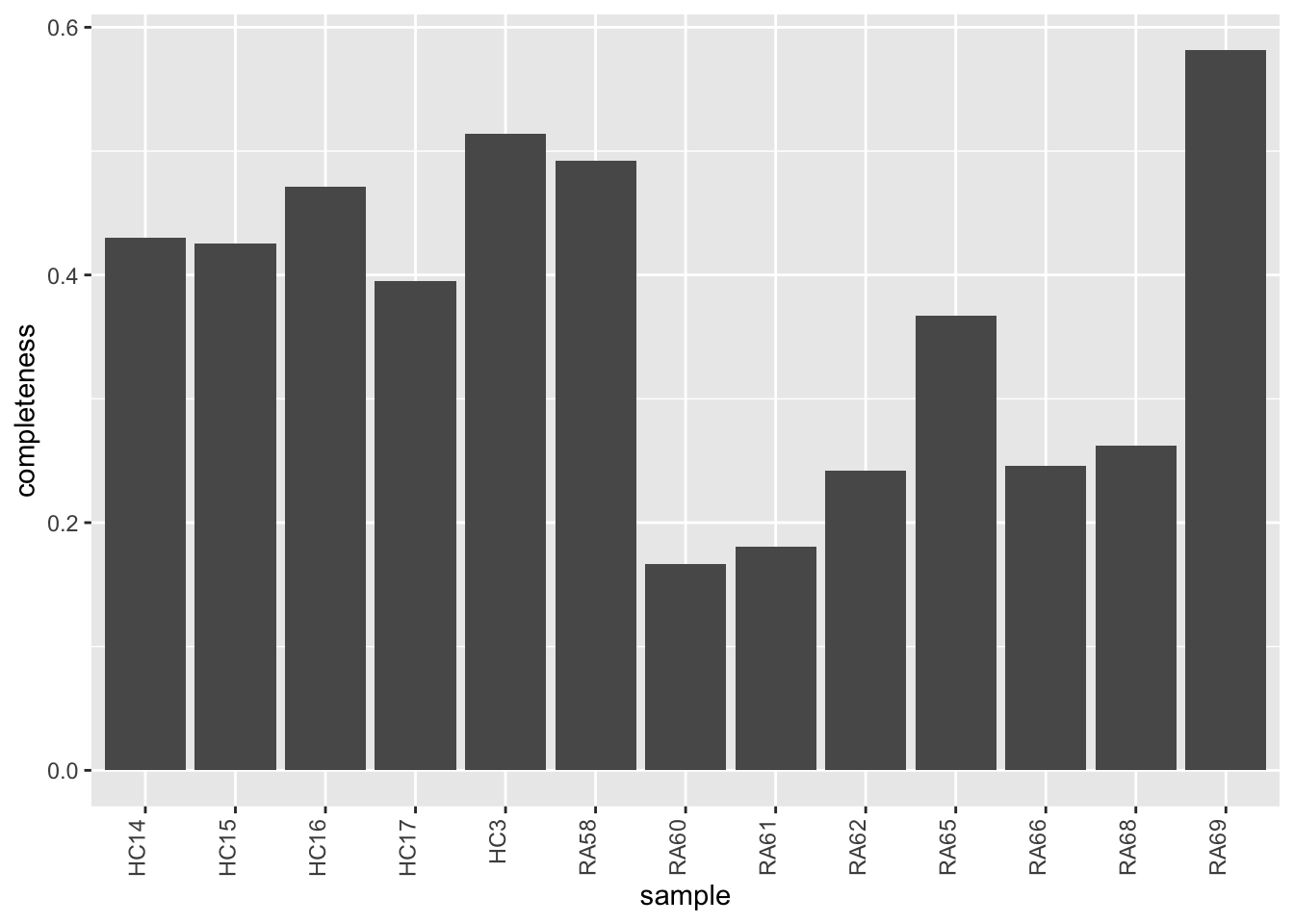
Plot a cumulative curve of missing value cut-off and remaining number of features
missRate <- tibble(id = rownames(countMat),
rate = rowSums(is.na(countMat))/ncol(countMat))
cumTab <- lapply(seq(0,1,0.05), function(cutRate) {
tibble(cut= cutRate,
per = sum(missRate$rate <= cutRate)/nrow(missRate))
} ) %>%
bind_rows()
ggplot(cumTab, aes(x=cut,y=per)) +
geom_line() +
xlab("Allowed missing value rate") +
ylab("Percentage of remaining features")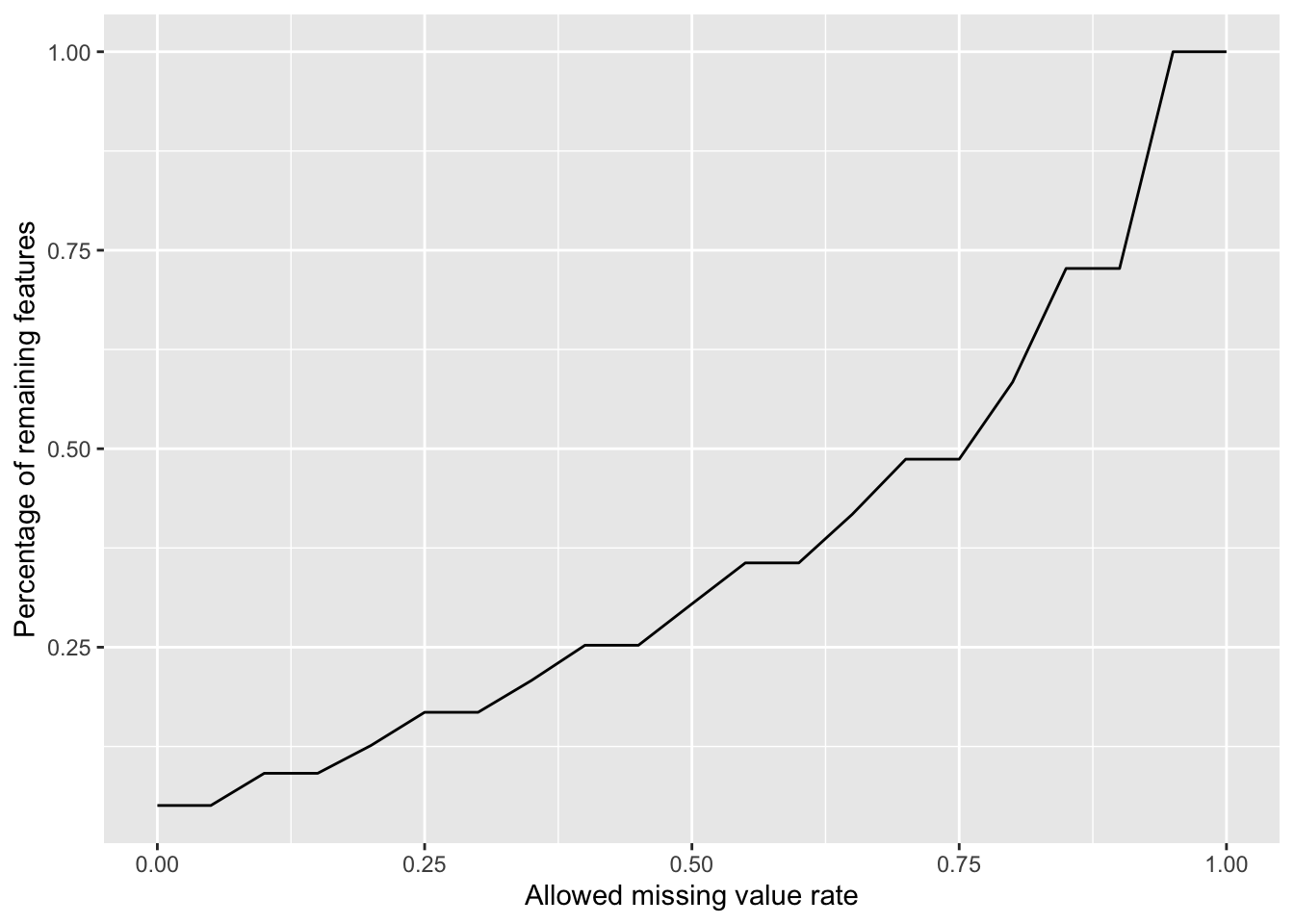 Missing value heatmap to check missing value structure
Missing value heatmap to check missing value structure
DEP::plot_missval(sePhos) Also looks very sparse.
Also looks very sparse.
Keep proteins detected in at least half of the sample (missing rate <= 0.5)
phosFilt <- sePhos[filter(missRate, rate <=0.5)$id,]
dim(phosFilt)[1] 2831 13Data distribution
countMat <- assay(phosFilt)
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value)) %>%
mutate(log2Val = log2(value))ggplot(countTab, aes(x=name, y=log2Val)) +
geom_boxplot() + geom_point()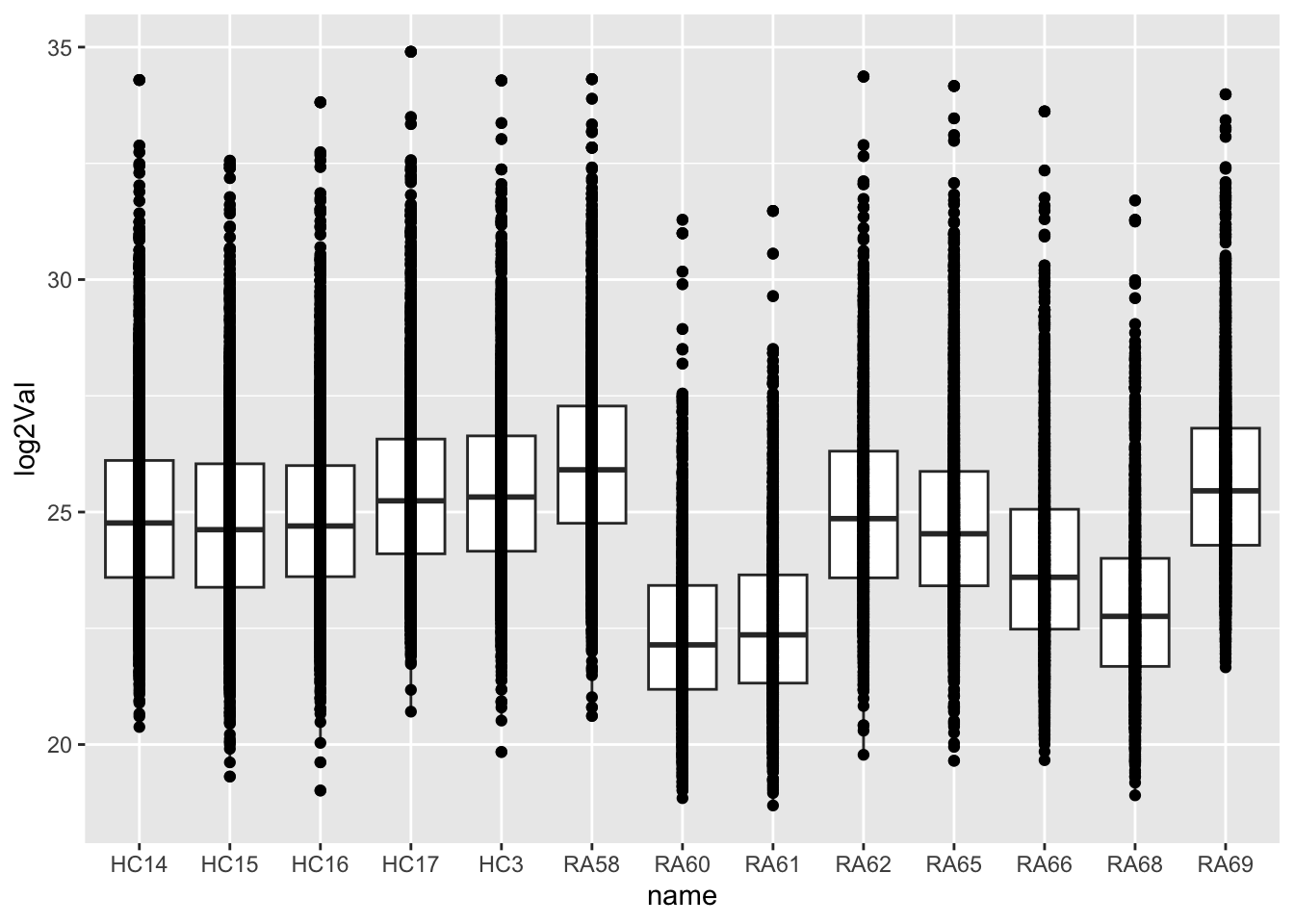 Was phospho-enrichment performed? There’s
strong difference in sample median RA60, 61, 68 are
using different buffer and with different startQuant
Was phospho-enrichment performed? There’s
strong difference in sample median RA60, 61, 68 are
using different buffer and with different startQuant
Imputation and normalization
Vst
protMat <- assay(phosFilt)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
phosNorm <- phosFilt
assay(phosNorm) <- normMatImputation
protImp <- DEP::impute(phosNorm, "QRILC")
assays(phosFilt)[["norm"]] <- normMat
assays(phosFilt)[["imputed"]] <- assay(protImp)
rowData(phosFilt) <- rowData(protImp)Distribution after normalizaiton
countMat <- assays(phosFilt)[["imputed"]]
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value))
ggplot(countTab, aes(x=name, y=value)) +
geom_boxplot() + geom_point()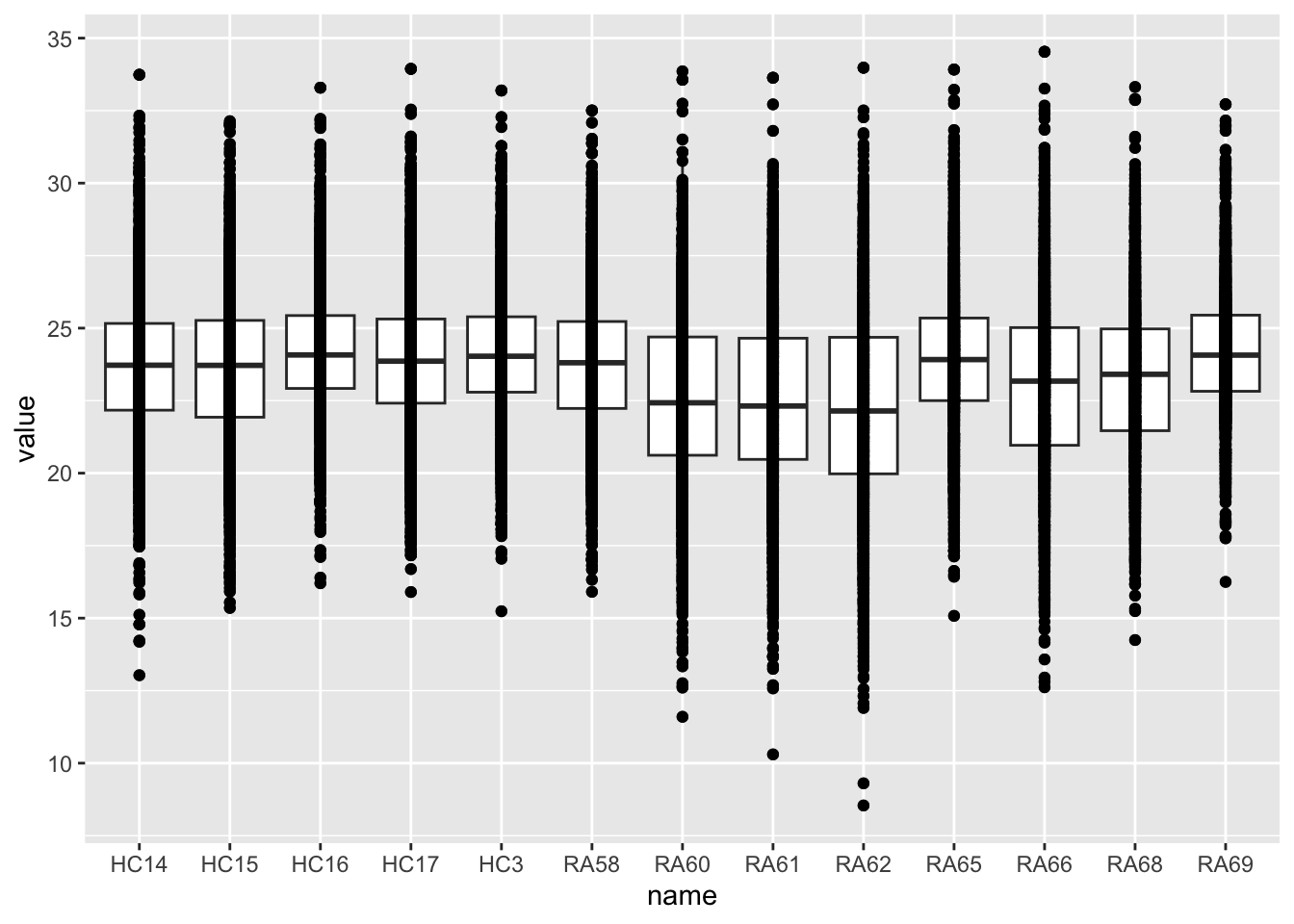
Mean versus variant plot
plotTab <- tibble(meanVal = rowMeans(countMat),
var = apply(countMat, 1, var))
ggplot(plotTab, aes(x=meanVal,y=var)) +
geom_point()
Heatmap visualization
library(pheatmap)
#select top 1000 most variant
colAnno <- colData(phosFilt) %>% data.frame()
colAnno <- colAnno[,c("Gender","group", "datePhos", "protConc", "quantStart", "sampleVol", "bufferComp")]
#colAnno[["sampleName"]] <- NULL
plotMat <- countMat[order(plotTab$var, decreasing = TRUE)[1:1000],]
pheatmap(plotMat, show_rownames = FALSE, scale = "row",
annotation_col = colAnno,
clustering_method = "ward.D2")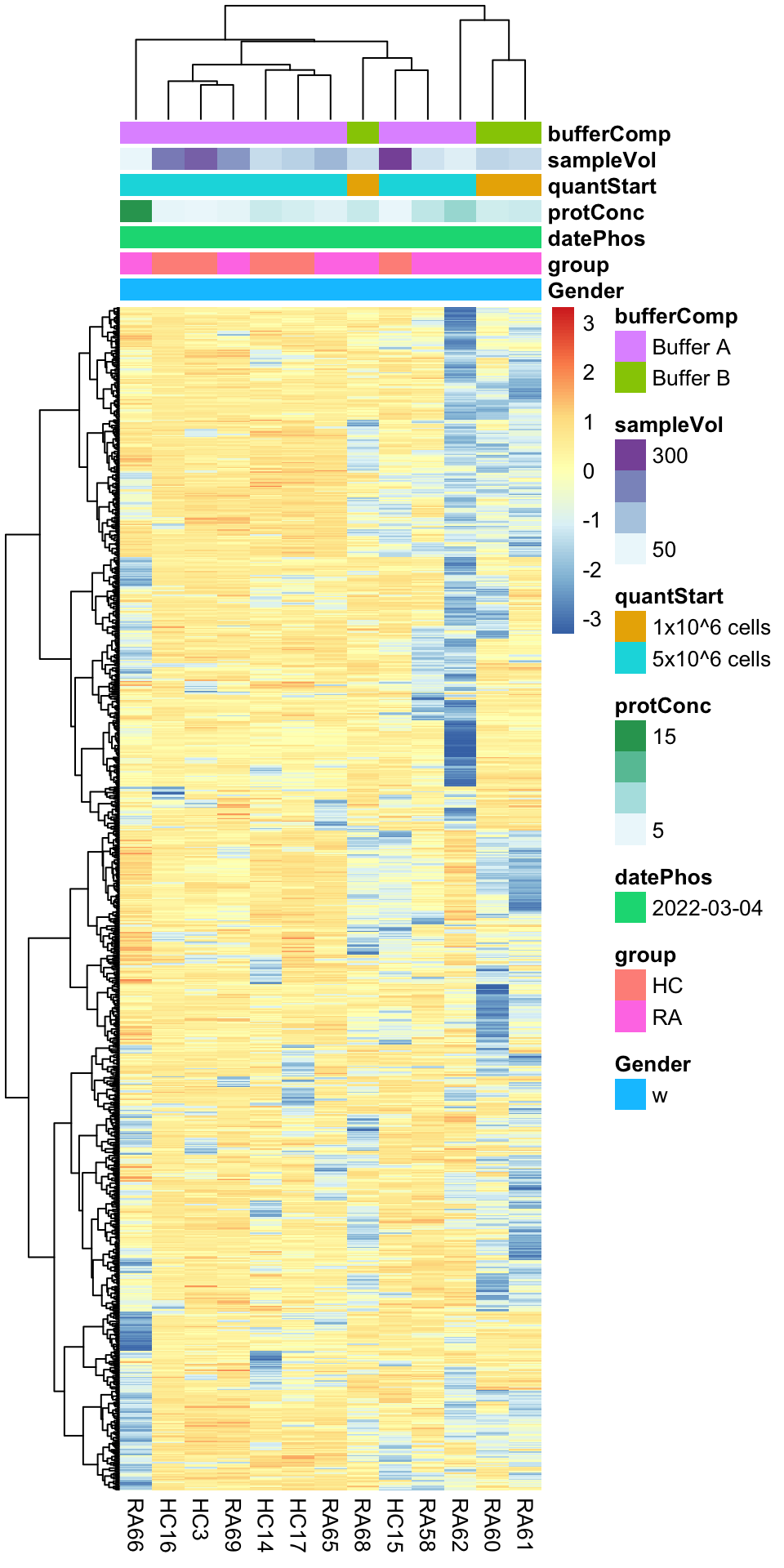
PCA
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))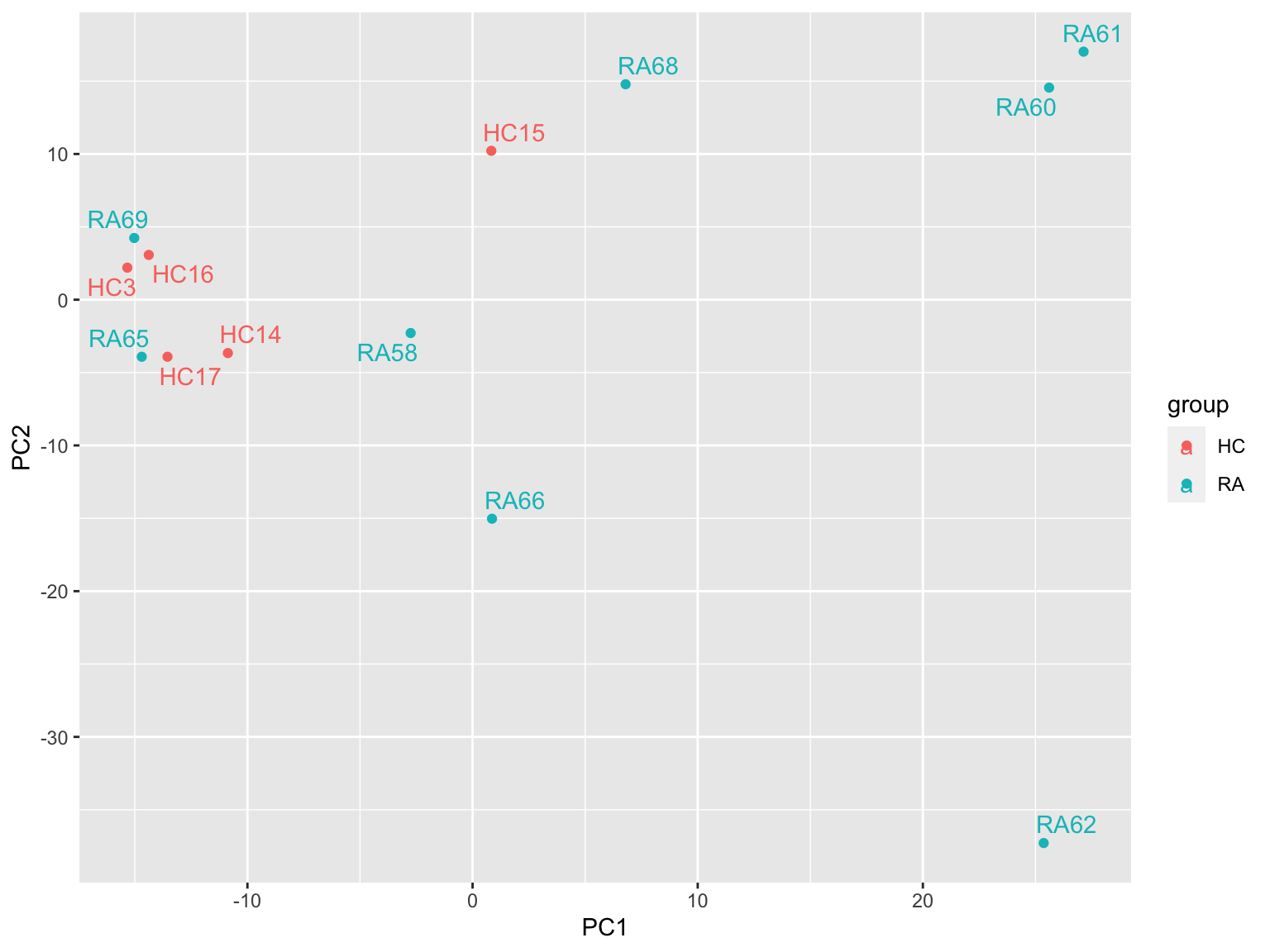 RA62 here may also be an outliers, although not as strong as
proteomics Similar to the proteomics data,
RA68,RA66,RA60 and RA61 are more separated to HC samples than other RA
samples
RA62 here may also be an outliers, although not as strong as
proteomics Similar to the proteomics data,
RA68,RA66,RA60 and RA61 are more separated to HC samples than other RA
samples
Colored by buffer composition
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = bufferComp)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))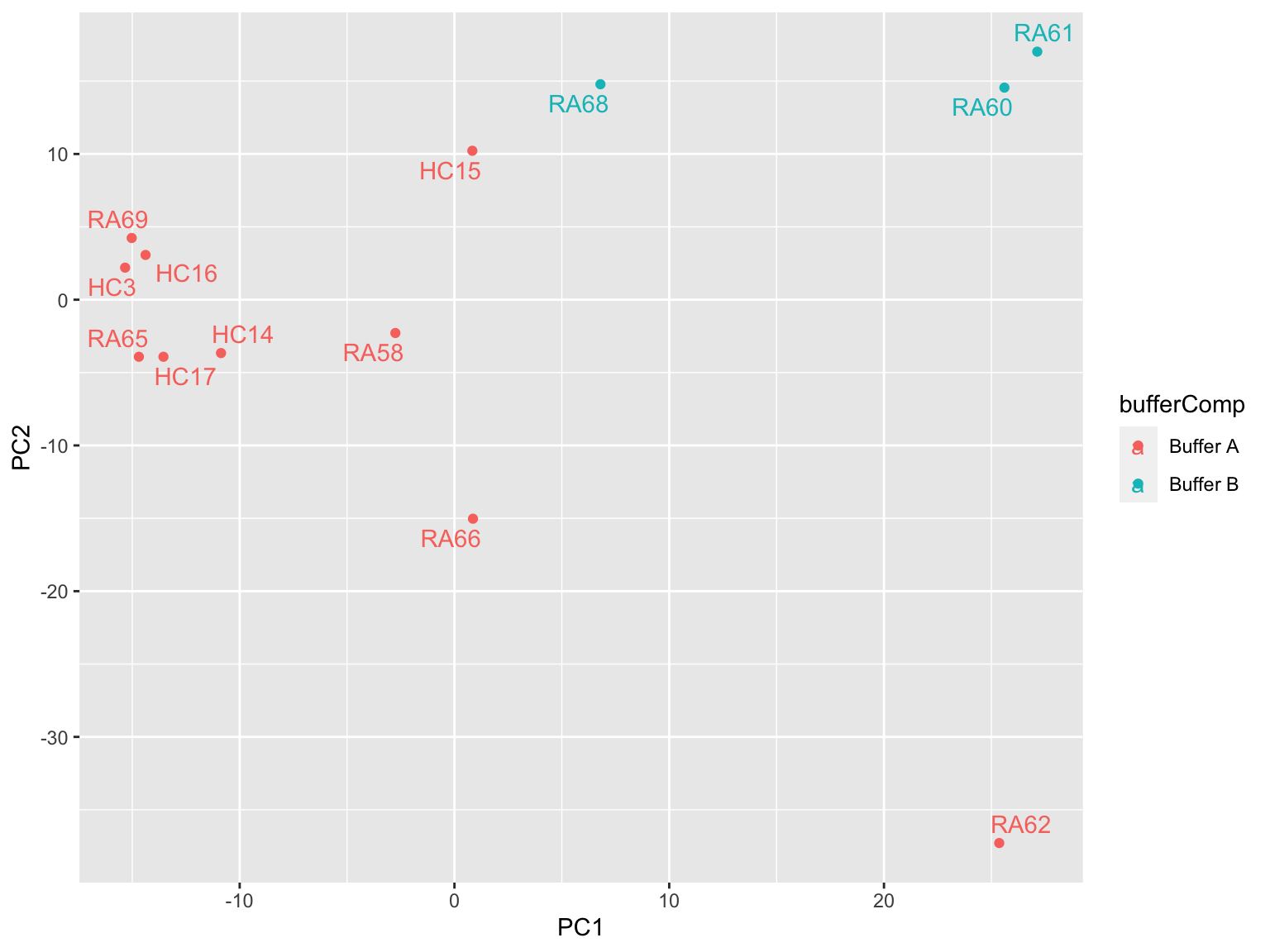
Remove one potential outlier, RA62 and redo preprocessing
phosSub <- phosFilt[,phosFilt$sampleID != "RA62"]Subset
phosSub <- sePhos[,sePhos$sampleID != "RA62"]
#remove proteins with more than 50% missing values
phosSub <- phosSub[rowSums(is.na(assay(phosSub)))/ncol(phosSub)<=0.5,]Vst
protMat <- assay(phosSub)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
phosNorm <- phosSub
assay(phosNorm) <- normMatImputation
protImp <- DEP::impute(phosNorm, "QRILC")
assays(phosSub)[["norm"]] <- normMat
assays(phosSub)[["imputed"]] <- assay(protImp)Redo pca
plotMat <- assays(phosSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))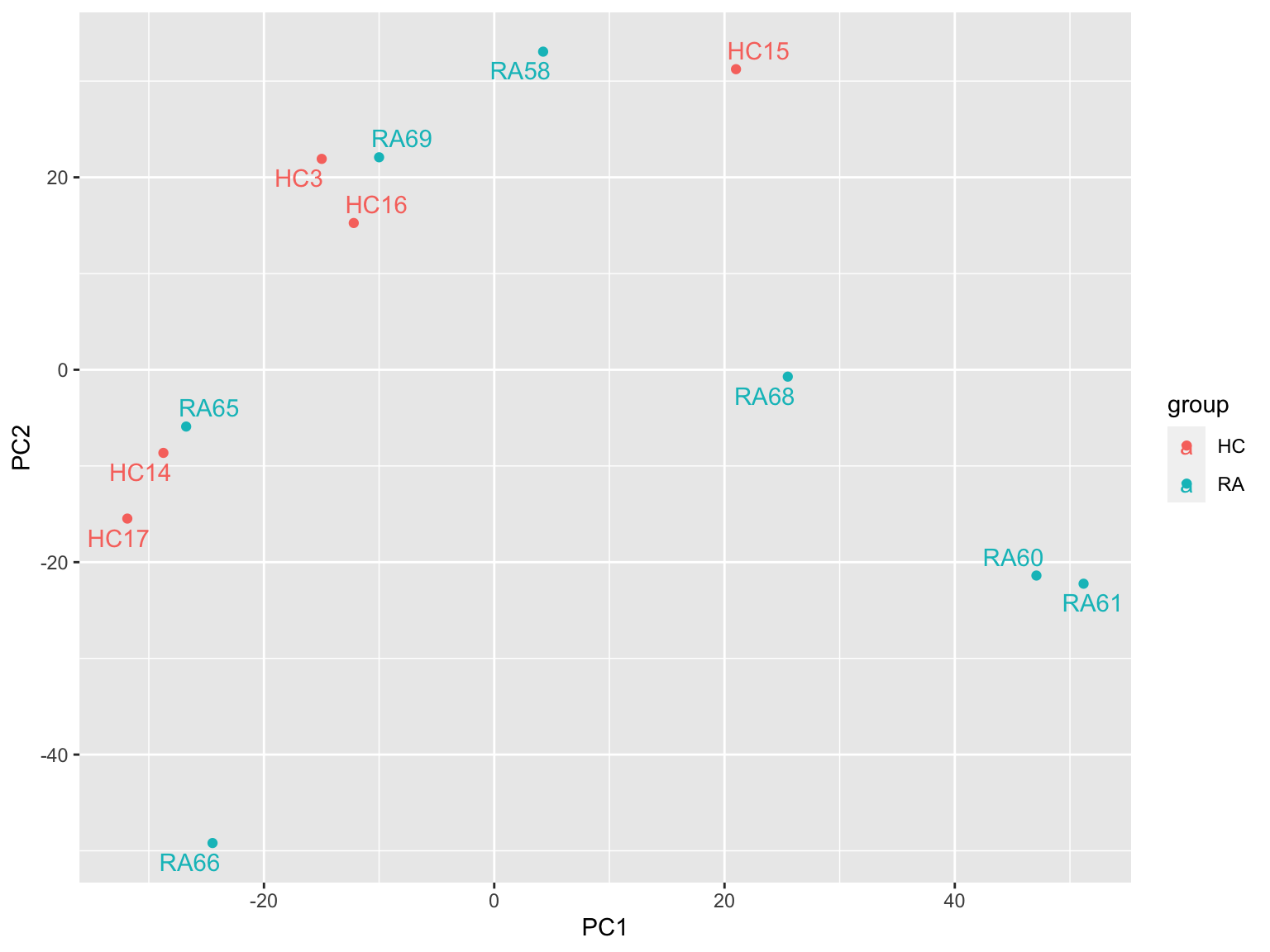
Colored by buffer composition
plotMat <- assays(phosSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = bufferComp)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))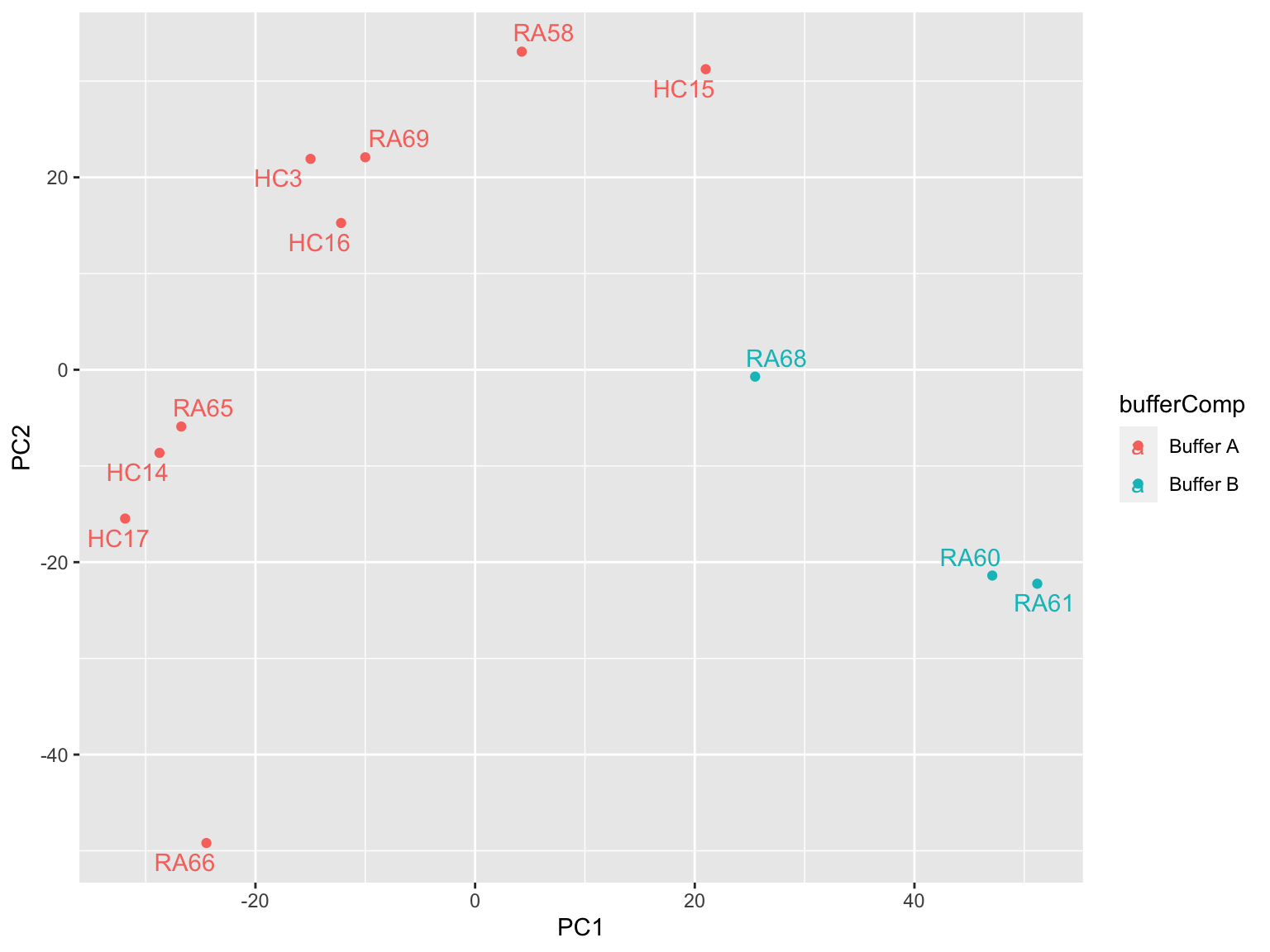
Phosphorylation normalized by protein expression
protBaseTab <- sumToTidy(protSub) %>%
select(colID, norm, name) %>%
group_by(colID, name) %>%
summarise(protNorm = mean(norm, na.rm=TRUE))
phosRatioTab <- sumToTidy(phosSub) %>%
select(rowID, colID, norm, name, site, symbol, bufferComp) %>%
left_join(protBaseTab, by = c("colID","name")) %>%
mutate(ratio = norm-protNorm) %>%
filter(!is.na(ratio))
seRatio <- tidyToSum(phosRatioTab, "rowID","colID","ratio", c("name","site","symbol"), annoCol = c("colID","bufferComp"))Methylation data
Use the processed methylation data from another script
load("../output/methData_20221118.RData")Filtering, only keep the top 5000 most variant genes for the multi-omics analysis
#remove genes on X,Y chromosome
methData <- methData[!seqnames(methData) %in% c("chrX","chrY"),]
methMat <- assays(methData)[["M"]]
#keep 5000 most variable CpG
sds <- genefilter::rowSds(methMat)
methMat <- methMat[order(sds, decreasing = T)[1:5000],]
colnames(methMat) <- methData$Sample_NameFACS data
facsTab <- readxl::read_xlsx("../data/Data_2023-02-16/FACS Data MFIs for Junyan.xlsx", sheet = 1) %>%
pivot_longer(-sample, names_to = "sampleName", values_to = "count") %>%
mutate(sampleName = str_remove(sampleName, "\\.{3}[:digit:]*")) %>%
dplyr::rename(feature = sample) %>%
mutate(feature =str_replace(feature, "memroy","memory"),
feature = str_replace(feature, "toal","total")) %>%
group_by(feature, sampleName) %>%
mutate(rep = seq_along(sampleName)) %>%
ungroup() %>%
mutate(rep = paste0("r",rep)) %>%
mutate(sampleID=ifelse(rep == "r1", sampleName, paste0(sampleName,"_",rep)),
count = as.numeric(count)) %>%
mutate(cell = str_extract(feature, "central memory|naive|effector memory|effector TEMRA|total")) %>%
mutate(marker = str_remove_all(str_remove(feature, cell)," "))
featureMap <- tibble(feature = unique(facsTab$feature)) %>%
mutate(id = paste0("f",seq_along(feature)))
facsTab <- left_join(facsTab, featureMap) %>%
left_join(patTab, by = c(sampleName = "sampleID"))
seFacs <- tidyToSum(facsTab, "id","sampleID","count",
annoRow = c("id","feature"),
annoCol = c("sampleID","sampleName","group", "dateFACS"))QC
plotTab <- facsTab
facsMat <- assay(seFacs)Measing value count
Per-feature
naSum <- group_by(facsTab, feature) %>%
summarise(numNA = sum(is.na(count))) %>%
arrange(desc(numNA)) %>%
mutate(feature = factor(feature, levels = feature))
ggplot(naSum, aes(x=feature, y=numNA)) +
geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))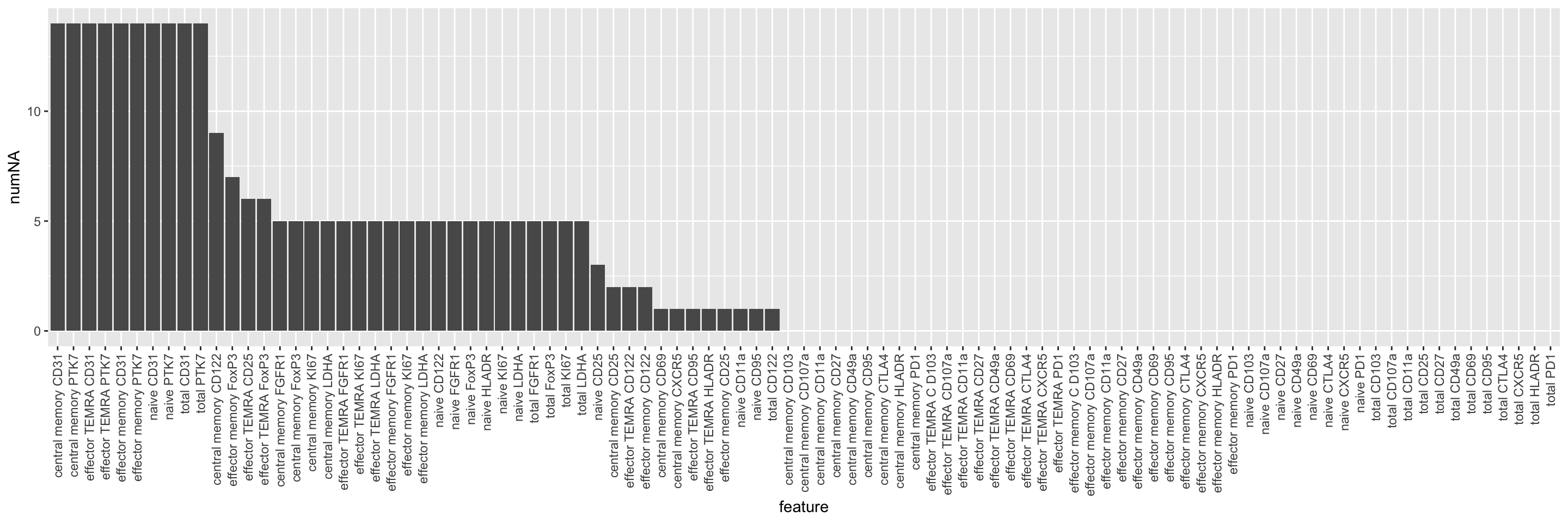
Per-sample
naSum <- group_by(facsTab, sampleID) %>%
summarise(numNA = sum(is.na(count))) %>%
arrange(desc(numNA)) %>%
mutate(sampleID = factor(sampleID, levels = sampleID))
ggplot(naSum, aes(x=sampleID, y=numNA)) +
geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))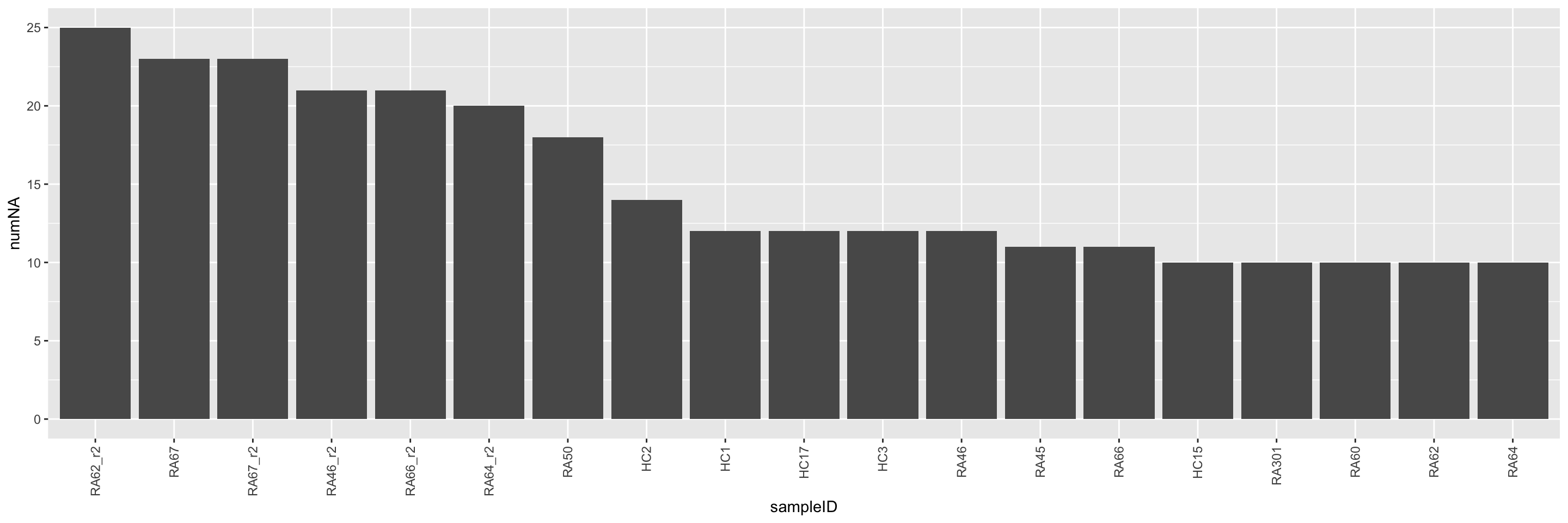
Missing value pattern
missTab <- facsTab %>% mutate(isNA = is.na(count))
ggplot(missTab, aes(x=marker, y=sampleID, fill=isNA)) +
geom_tile() +
facet_wrap(~cell, nrow = 1) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))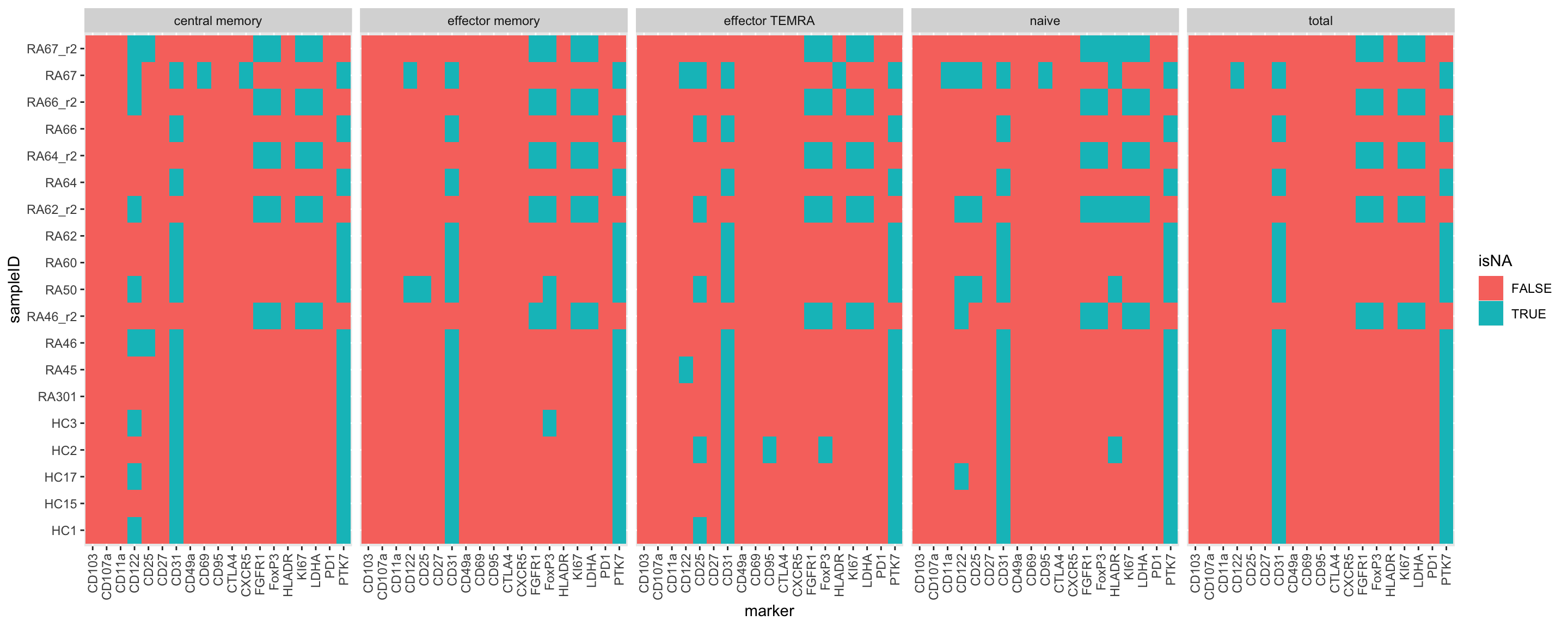 Replicate 2 uses a different panel?
Replicate 2 uses a different panel?
Feature distribution
Per-sample
Raw scale
ggplot(plotTab, aes(x=sampleID, y=count, fill = group)) +
geom_boxplot() + geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))
glog transformed
ggplot(plotTab, aes(x=sampleID, y=glog2(count), fill = group)) +
geom_boxplot() + geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))
Per feature
Raw scale
ggplot(plotTab, aes(x=feature, y=count)) +
geom_boxplot() + geom_point(aes(col=group)) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))
glog transformed
ggplot(plotTab, aes(x=feature, y=glog2(count))) +
geom_boxplot() + geom_point(aes(col= group)) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust =0.5))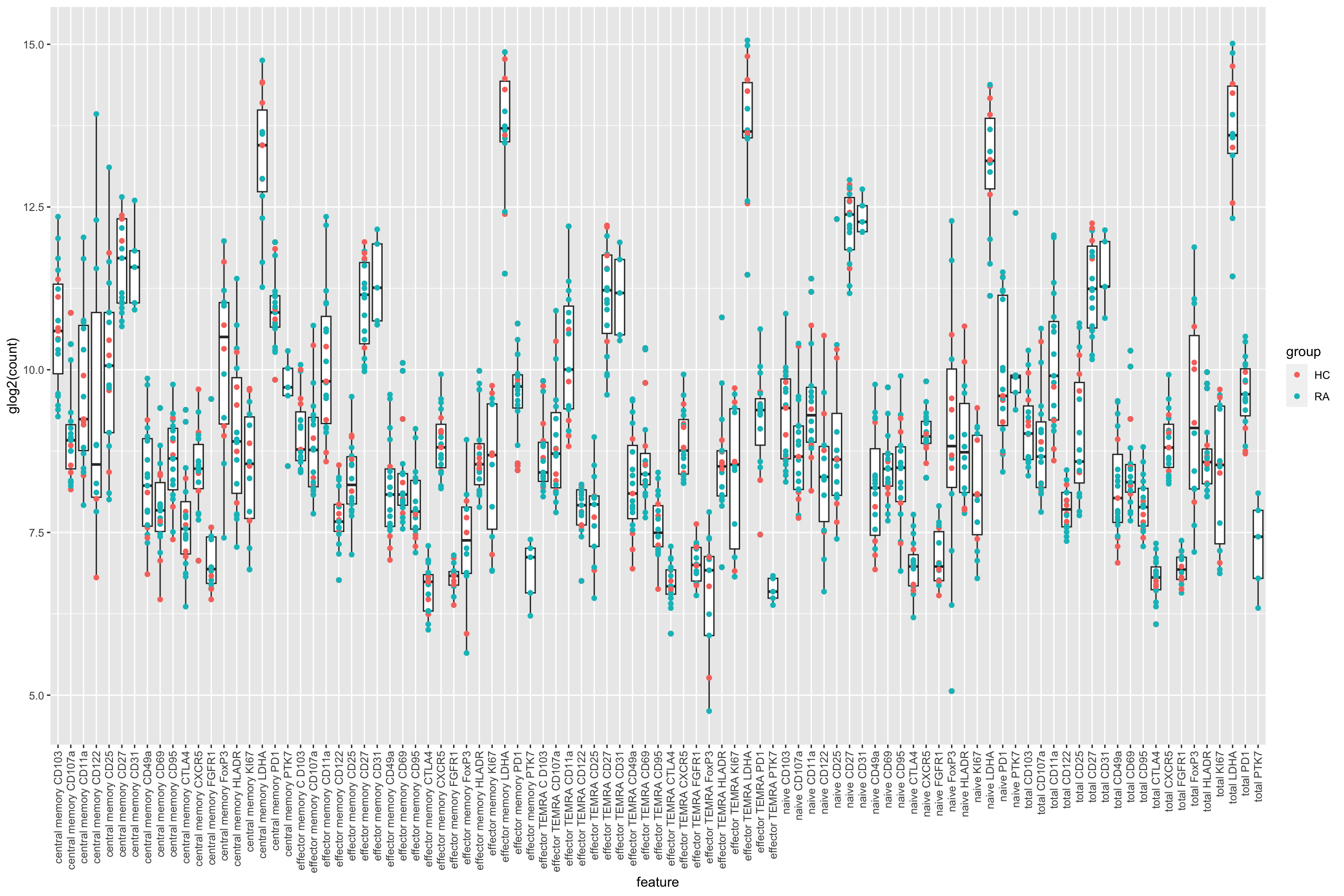
PCA
Raw scale
facsMat <- assay(seFacs)
#only use features with complete measurement
facsMat <- facsMat[complete.cases(facsMat),]
pcRes <- prcomp(t(facsMat), scale. = TRUE, center = TRUE)
plotTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
mutate(group = seFacs[,sampleID]$group)
ggplot(plotTab, aes(x=PC1, y=PC2, col = group, label = sampleID)) +
geom_point() +
geom_text()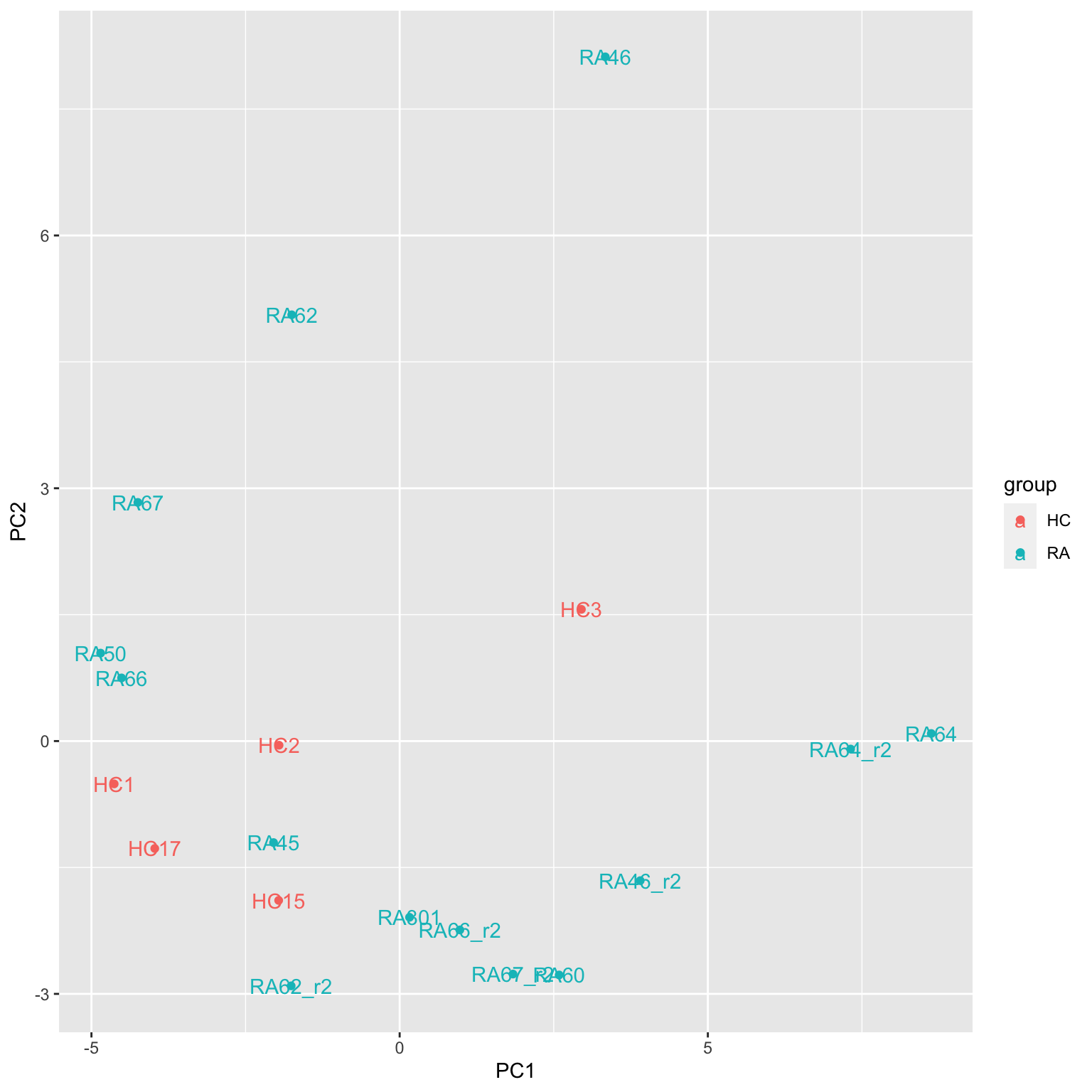
glog transformed
Colored by phenotype
facsMat <- jyluMisc::glog2(facsMat)
pcRes <- prcomp(t(facsMat), scale. = TRUE, center = TRUE)
plotTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
mutate(group = seFacs[,sampleID]$group)
ggplot(plotTab, aes(x=PC1, y=PC2, col = group, label = sampleID)) +
geom_point() +
geom_text()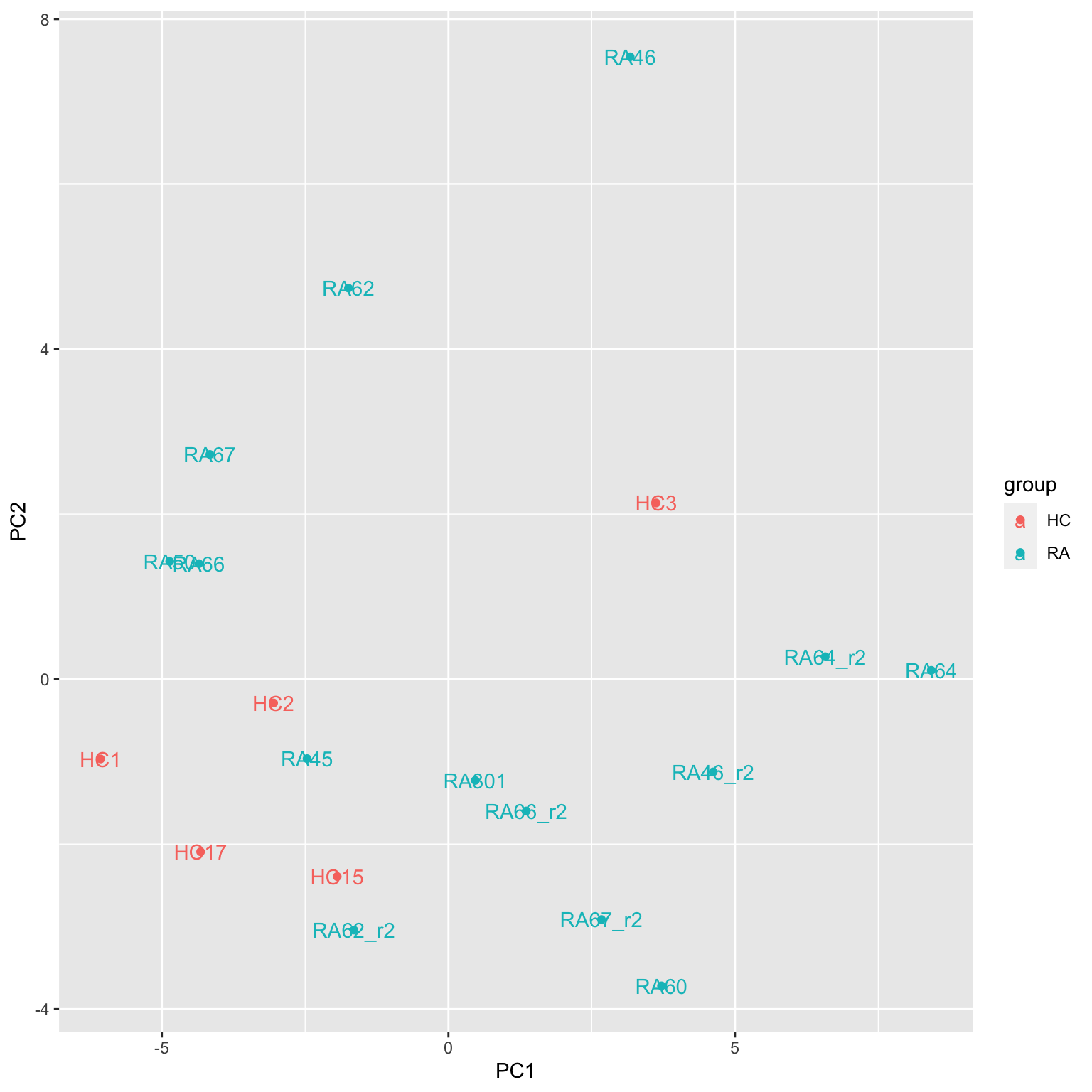 Replicates look very different
Replicates look very different
Are replicates all measured on a different date?
Compare reproducibility among replicates
repSmp <- unique(filter(facsTab, rep == "r2")$sampleName)
comTab <- filter(facsTab, sampleName %in% repSmp) %>%
select(feature, sampleName, rep, count) %>%
mutate(count = glog2(count)) %>%
pivot_wider(names_from = rep, values_from = count)
ggplot(comTab, aes(x=r1, y=r2)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, color = "red")+
facet_wrap(~sampleName) +
xlim(-1,15) + ylim(-1,15)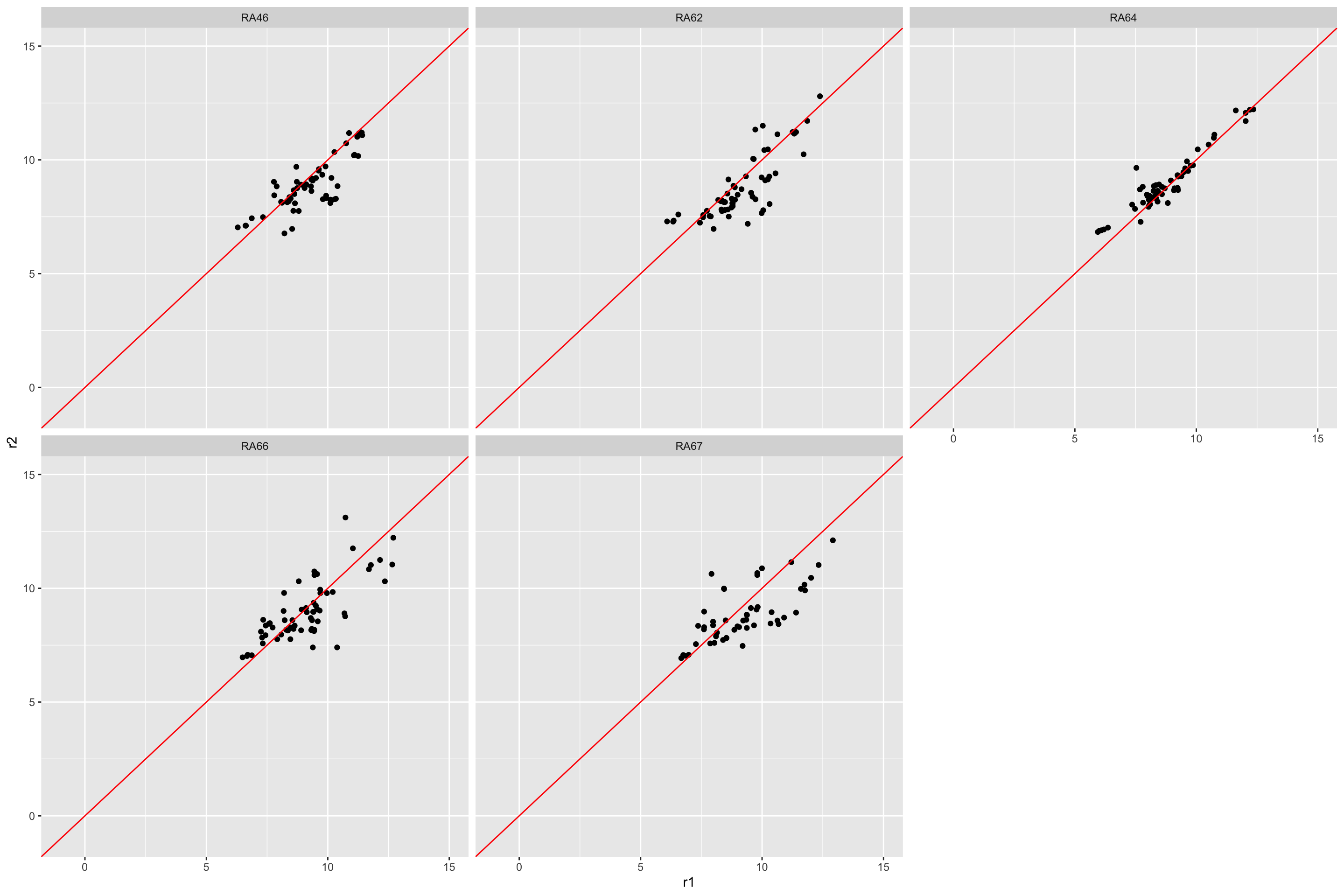
Remove R2 samples
seFacs <- seFacs[,!str_detect(seFacs$sampleID,"r2")]
seFacs$sampleID <- seFacs$sampleName
seFacs$sampleName <- NULLRemove feature without any measurment
seFacs <- seFacs[rowSums(!is.na(assay(seFacs)))>0,]Cell population
popTab <- readxl::read_xlsx("../data/Data_2023-02-16/FACS Data MFIs for Junyan.xlsx", sheet = 2) %>%
pivot_longer(-`...1`, names_to = "sampleName", values_to = "count") %>%
mutate(sampleName = str_remove(sampleName, "\\.{3}[:digit:]*")) %>%
dplyr::rename(feature = `...1`) %>%
group_by(feature, sampleName) %>%
mutate(rep = seq_along(sampleName)) %>%
ungroup() %>%
mutate(rep = paste0("r",rep)) %>%
mutate(sampleID=ifelse(rep == "r1", sampleName, paste0(sampleName,"_",rep)),
count = as.numeric(count))
popTab <- popTab %>%
left_join(patTab, by = c(sampleName = "sampleID"))ggplot(popTab, aes(x=group, y=count, label = sampleID)) +
geom_boxplot() +
geom_point(aes(col = group)) +
ggrepel::geom_text_repel(max.overlaps = Inf) +
facet_wrap(~feature, scale = "free")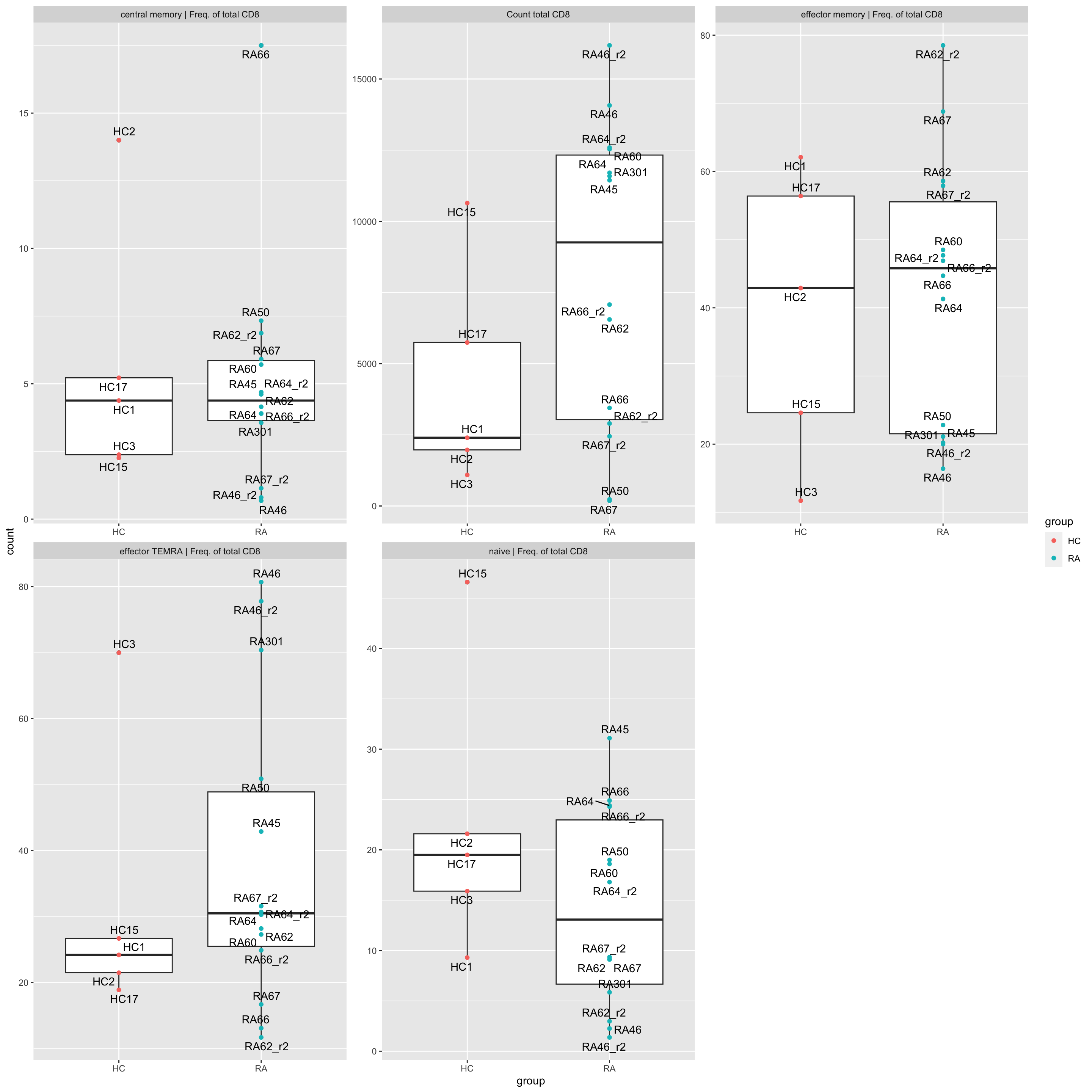 May not be very informative, so it will not be used for
multi-omic analysis
May not be very informative, so it will not be used for
multi-omic analysis
Process new DIA Proteomics data received in March, 2024
Read sample metadata
smpMeta <- readxl::read_xlsx("../data/TF0837_results/2023-12-18_SampleSubmission_Kraus_post randomization.xlsx") %>%
dplyr::rename(id = `Sample Number (same as vial label)`,
sampleID = `Sample Name`,
quantStart = "Quantity of Starting Material") %>%
mutate(quantStart = str_remove(quantStart," lymphocytes")) %>%
separate(quantStart, into = c("baseN","expN"), sep = "\\*10\\^") %>%
mutate(quantStart = as.numeric(baseN)*10^as.numeric(expN)) %>%
select(id, sampleID, quantStart)Read data table
protTab <- readxl::read_xlsx("../data/TF0837_results/TF0837_filtered_proteinGroups.xlsx") %>%
select(`PG.ProteinGroups`, `PG.Genes`, contains("PG.Quantity")) %>%
dplyr::rename(name = "PG.ProteinGroups", symbol = "PG.Genes") %>%
pivot_longer(-c(name, symbol), names_to = "id", values_to = "count") %>%
mutate(id = as.numeric(str_extract(id, "(?<=TF0837-)..(?=\\.raw)"))) %>%
filter(count>0) %>% left_join(smpMeta, by = "id") %>%
left_join(patTab, by = "sampleID")
idMap <- unique(protTab$name)
idMap <- structure(paste0("prot",seq_along(idMap)), names = idMap)
protTab <- mutate(protTab, ID = idMap[name])Choose the first symbol if multiple symbols are present in the symbol column
# Get the last symbol of a protein that has multiple gene symbols
getOneSymbol <- function(Gene) {
outStr <- sapply(Gene, function(x) {
sp <- str_split(x, ";")[[1]]
sp[length(sp)]
})
names(outStr) <- NULL
outStr
}
protTab$symbol <- getOneSymbol(protTab$symbol)
#only keep proteins with symbols
protTab <- filter(protTab, !symbol %in% c("",NA), !str_detect(symbol, "SWISS-PROT"))Created summarised experiment
seDIA <- jyluMisc::tidyToSum(protTab, "ID", "sampleID","count",
annoRow = c("ID","name","symbol"),
annoCol = c(colnames(patTab),"quantStart"))Some samples don’t have the group information, but this can be determined by the sample name
seDIA$group <- ifelse(is.na(seDIA$group), ifelse(str_detect(colnames(seDIA),"HC"), "HC", "RA"), seDIA$group)QA
Data completeness per sample
countMat <- assay(seDIA)
plotTab <- tibble(sample = colnames(seDIA),
perNA = colSums(is.na(countMat))/nrow(countMat),
startQuant = seDIA$quantStart)
ggplot(plotTab, aes(x=sample, y=1-perNA)) +
geom_bar(stat = "identity", aes(fill = startQuant)) +
ylab("completeness") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust=0))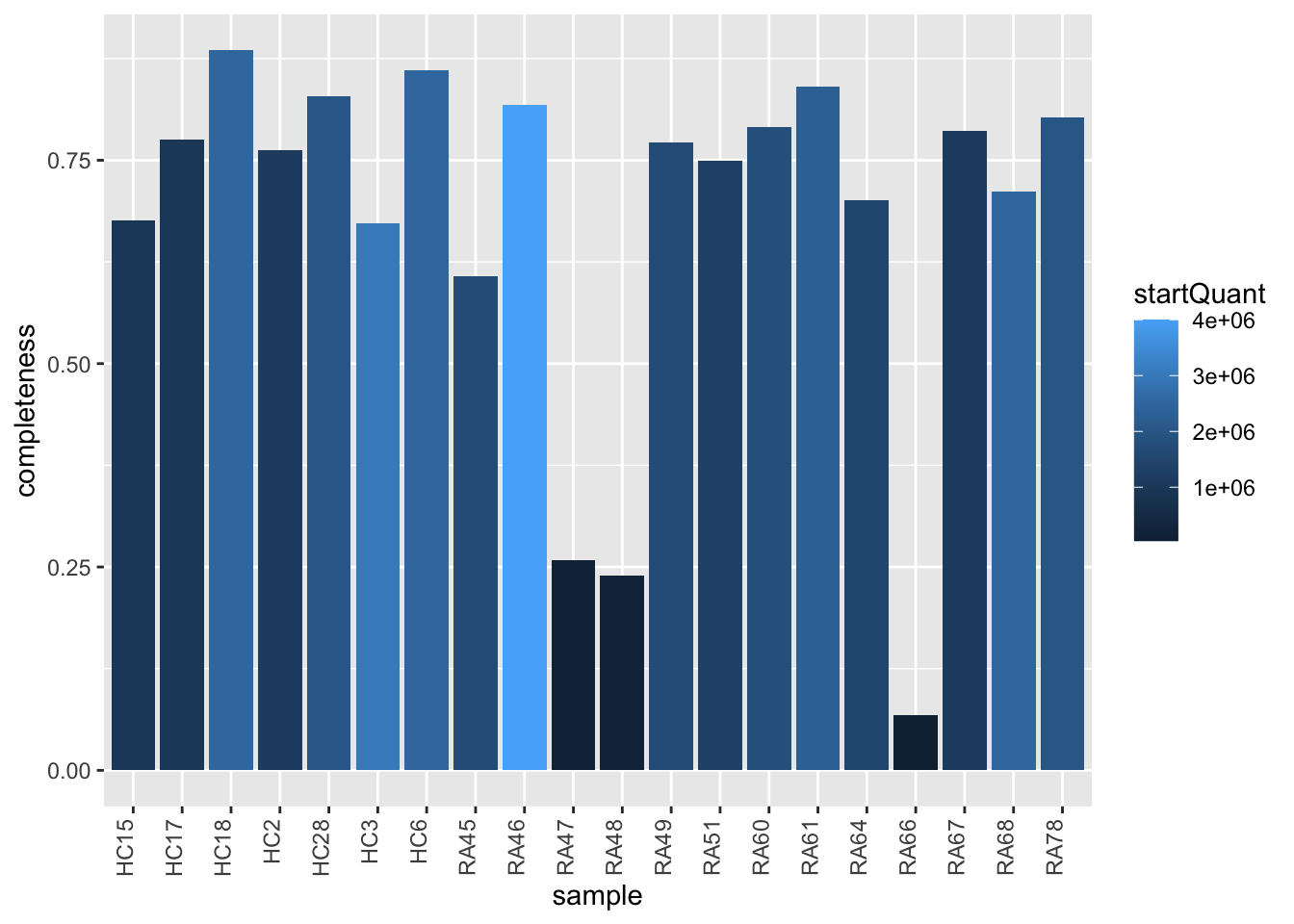
Plot a cumulative curve of missing value cut-off and remaining number of features
missRate <- tibble(id = rownames(countMat),
rate = rowSums(is.na(countMat))/ncol(countMat))
cumTab <- lapply(seq(0,1,0.05), function(cutRate) {
tibble(cut= cutRate,
per = sum(missRate$rate <= cutRate)/nrow(missRate))
} ) %>%
bind_rows()
ggplot(cumTab, aes(x=cut,y=per)) +
geom_line() +
xlab("Allowed missing value rate") +
ylab("Percentage of remaining features")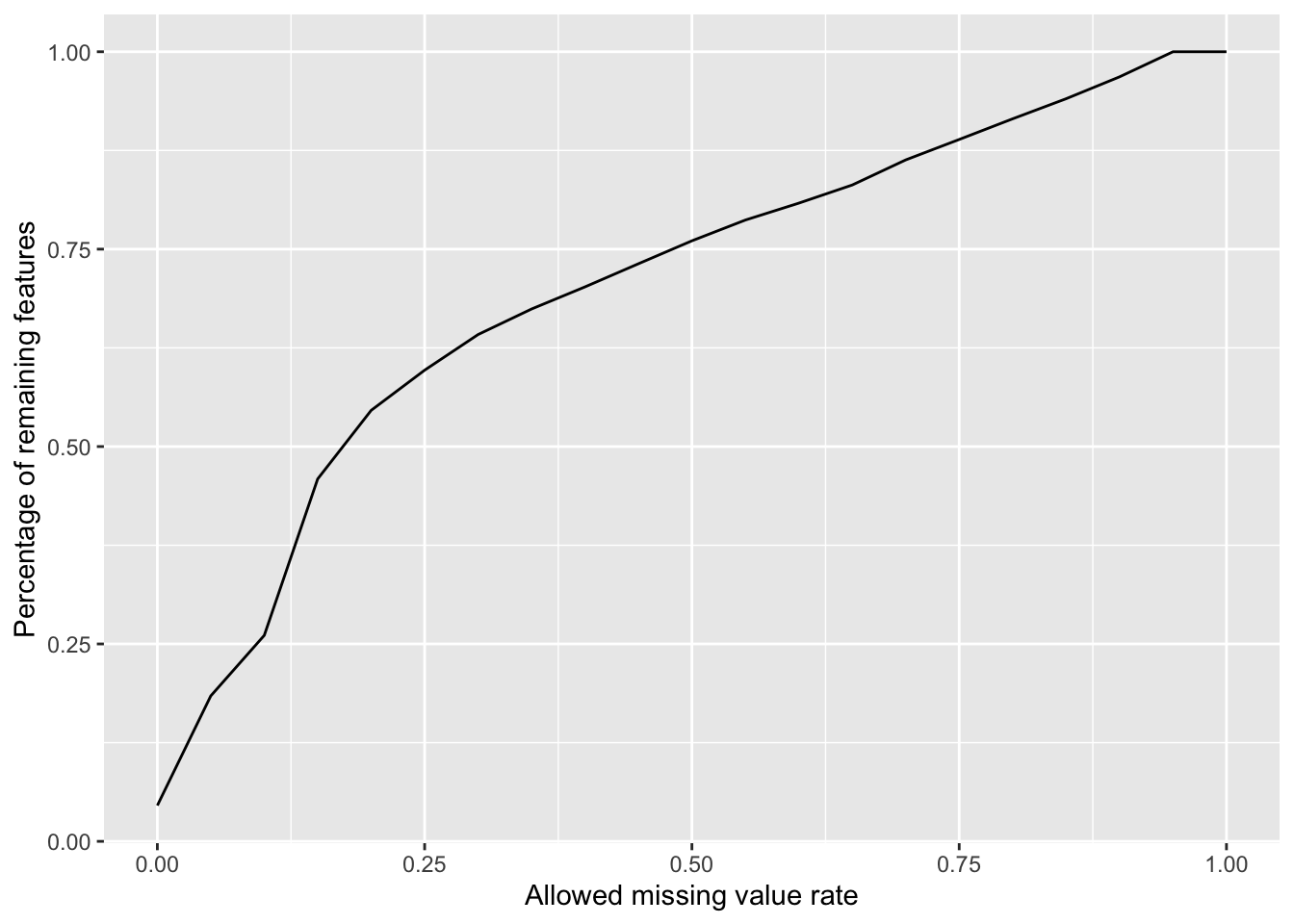 Missing value heatmap to check missing value structure
Missing value heatmap to check missing value structure
Visualize the missing value pattern
DEP::plot_missval(seDIA) Looks pretty sparse, maybe due to the DDA data aquisition method
Looks pretty sparse, maybe due to the DDA data aquisition method
Keep proteins detected in at least half of the sample (missing rate <= 0.5)
protFilt <- seDIA[filter(missRate, rate <=0.5)$id,]
dim(protFilt)[1] 5292 20Data distribution
countMat <- assay(protFilt)
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value)) %>%
mutate(log2Val = log2(value))ggplot(countTab, aes(x=name, y=log2Val)) +
geom_boxplot() + geom_point()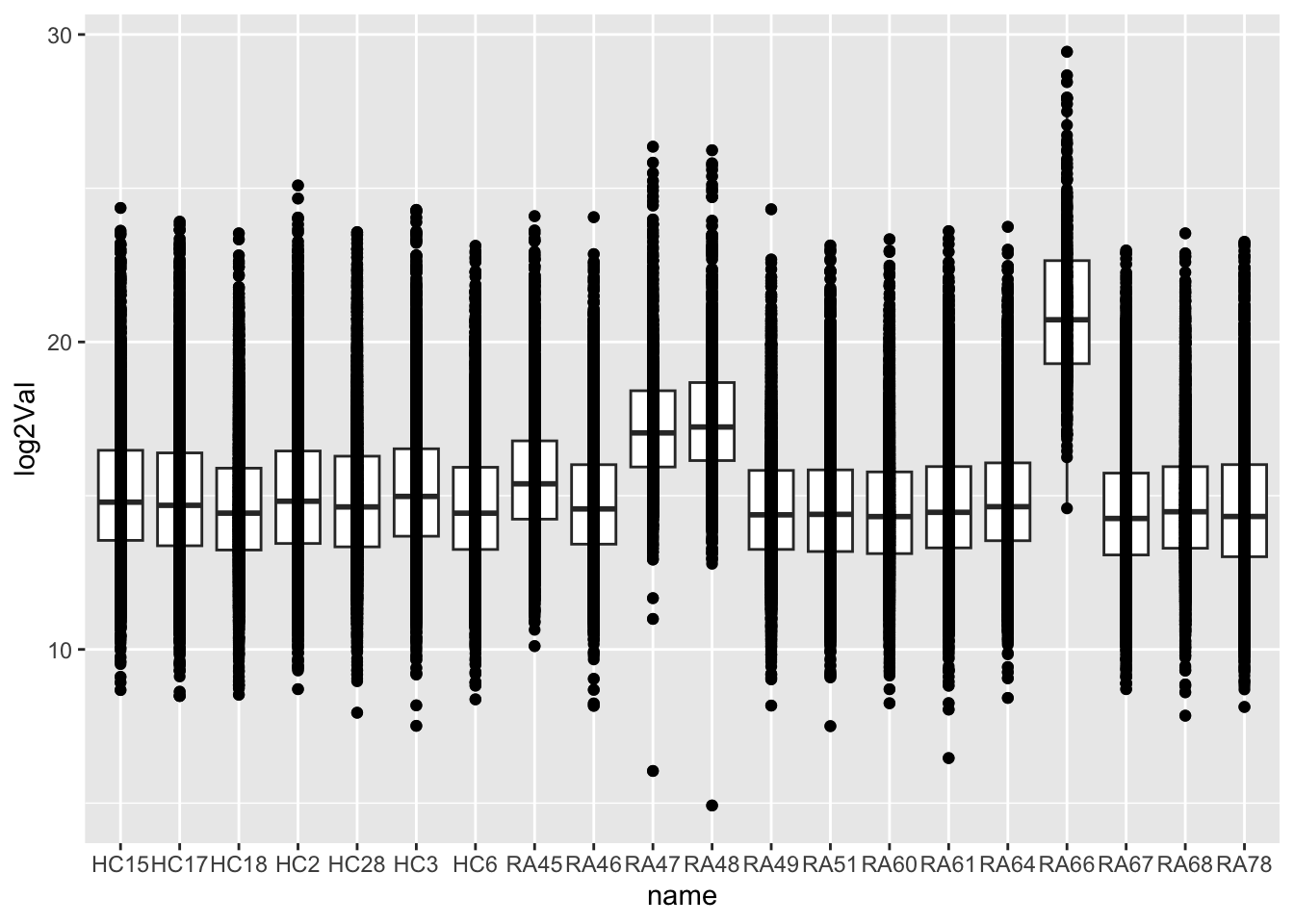
Imputation and normalization
Vst
protMat <- assay(protFilt)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
protNorm <- protFilt
assay(protNorm) <- normMatImputation
protImp <- DEP::impute(protNorm, "QRILC")
assays(protFilt)[["norm"]] <- normMat
assays(protFilt)[["imputed"]] <- assay(protImp)
rowData(protFilt) <- rowData(protImp)Distribution after normalizaiton
countMat <- assays(protFilt)[["imputed"]]
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value))
ggplot(countTab, aes(x=name, y=value)) +
geom_boxplot() + geom_point()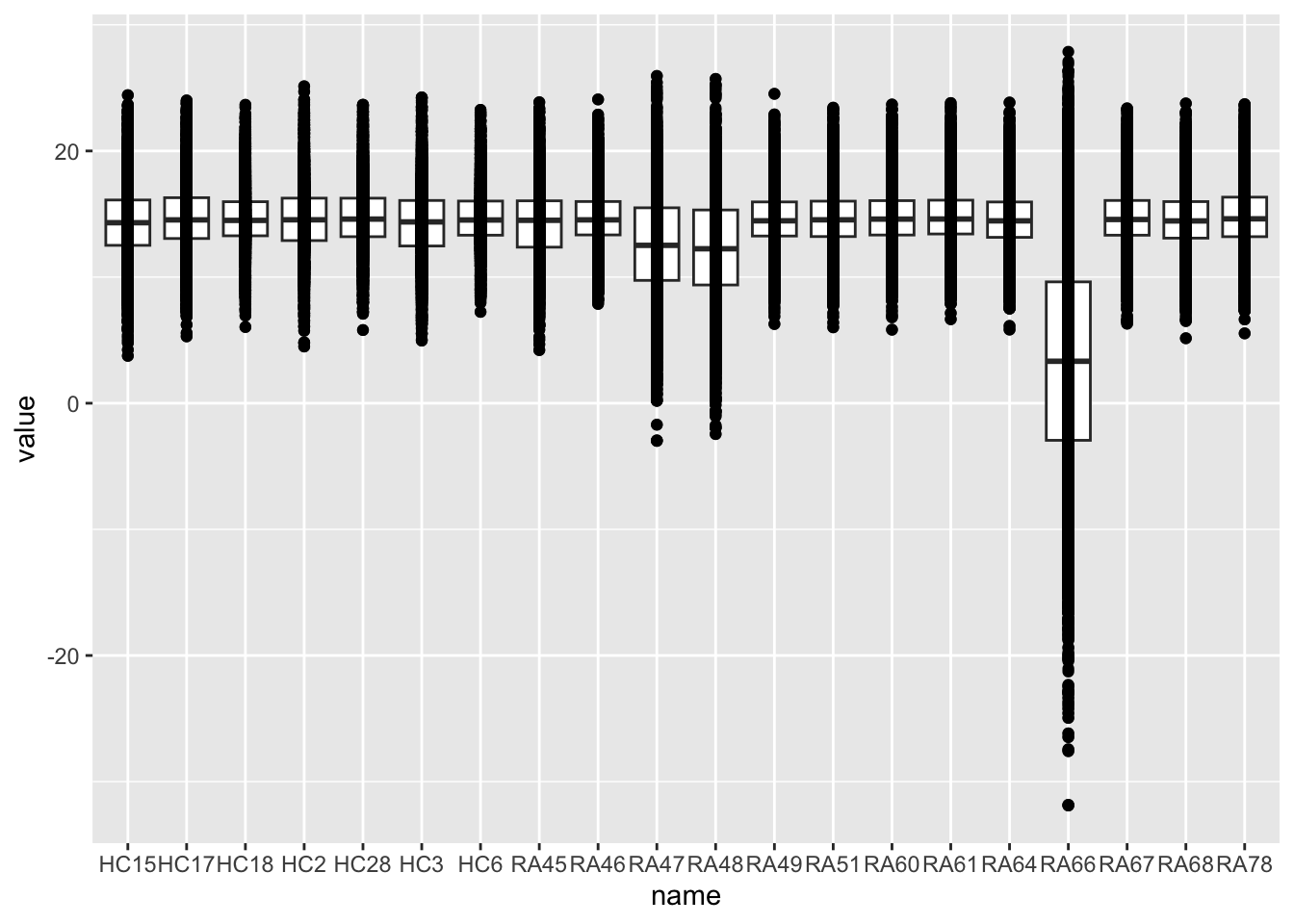
Mean versus variant plot
plotTab <- tibble(meanVal = rowMeans(countMat),
var = apply(countMat, 1, var))
ggplot(plotTab, aes(x=meanVal,y=var)) +
geom_point()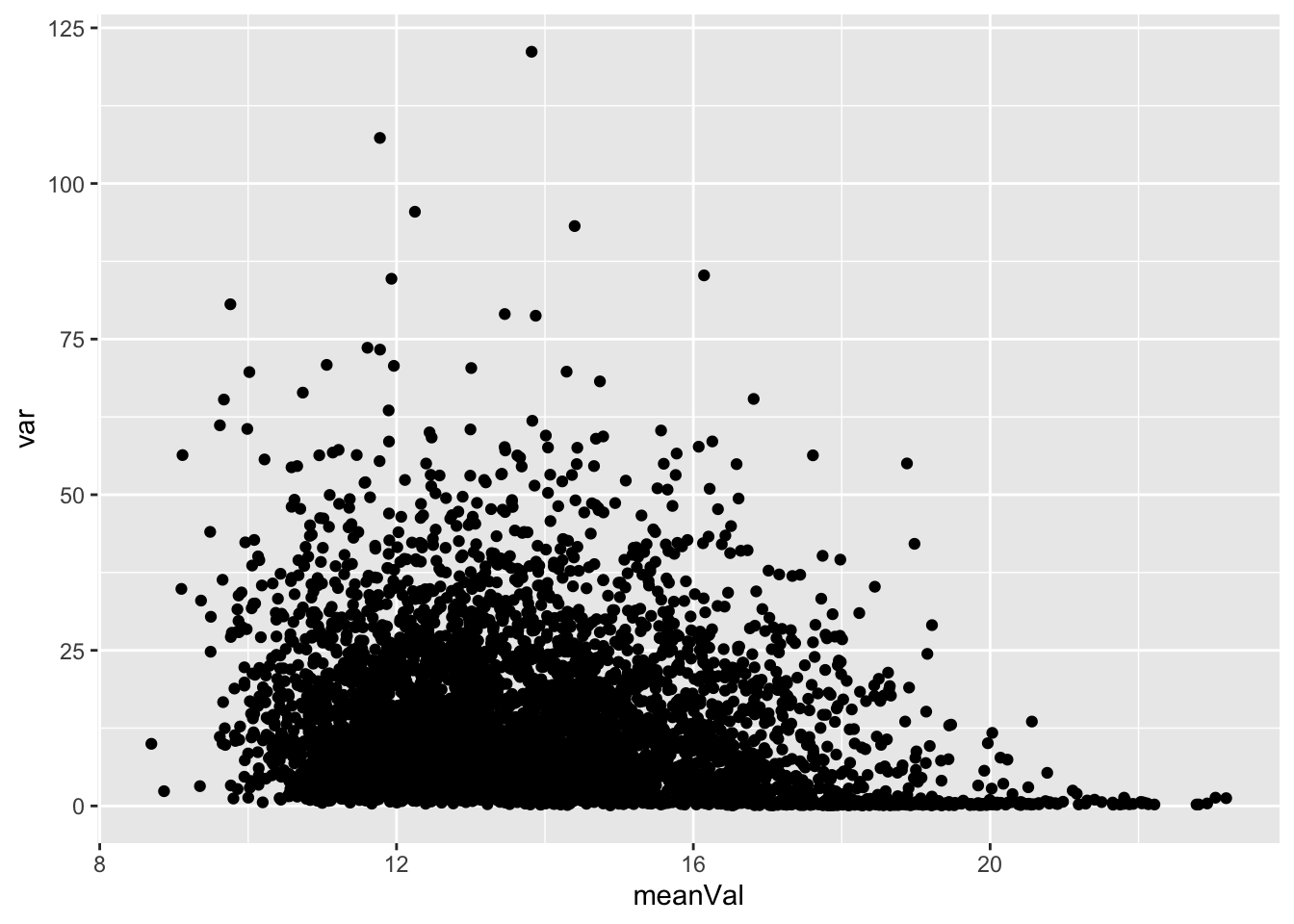
Heatmap visualization
library(pheatmap)
#select top 1000 most variant
colAnno <- colData(protFilt) %>% data.frame()
colAnno <- colAnno[,c("Gender","group","quantStart")]
#colAnno[["sampleName"]] <- NULL
plotMat <- countMat[order(plotTab$var, decreasing = TRUE)[1:1000],]
pheatmap(plotMat, show_rownames = FALSE, scale = "row",
annotation_col = colAnno,
clustering_method = "ward.D2")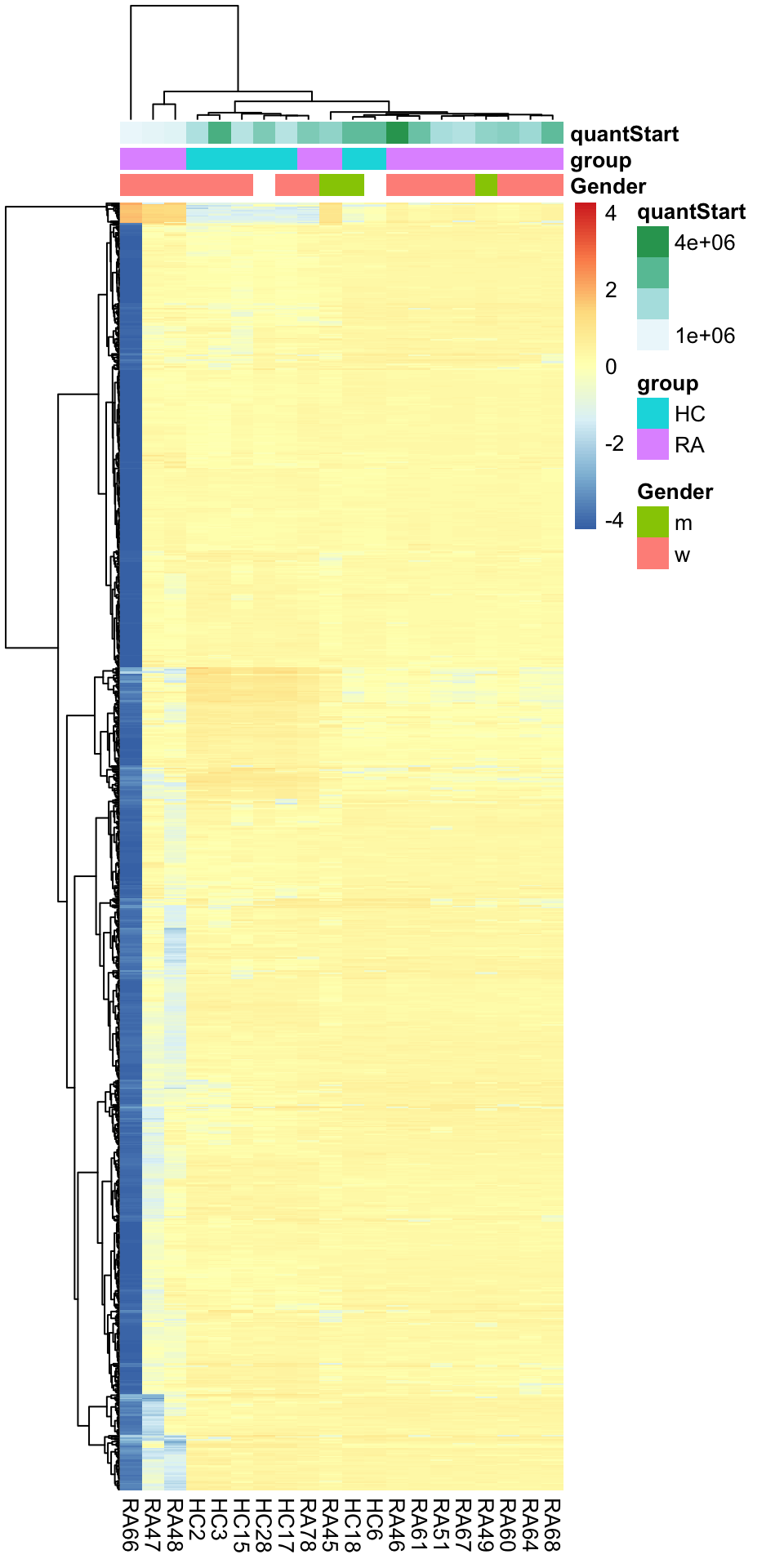 Buffer composition and startQuant could be potential counfunding
factor
Buffer composition and startQuant could be potential counfunding
factor
PCA
prRes <- prcomp(t(plotMat), scale. = FALSE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))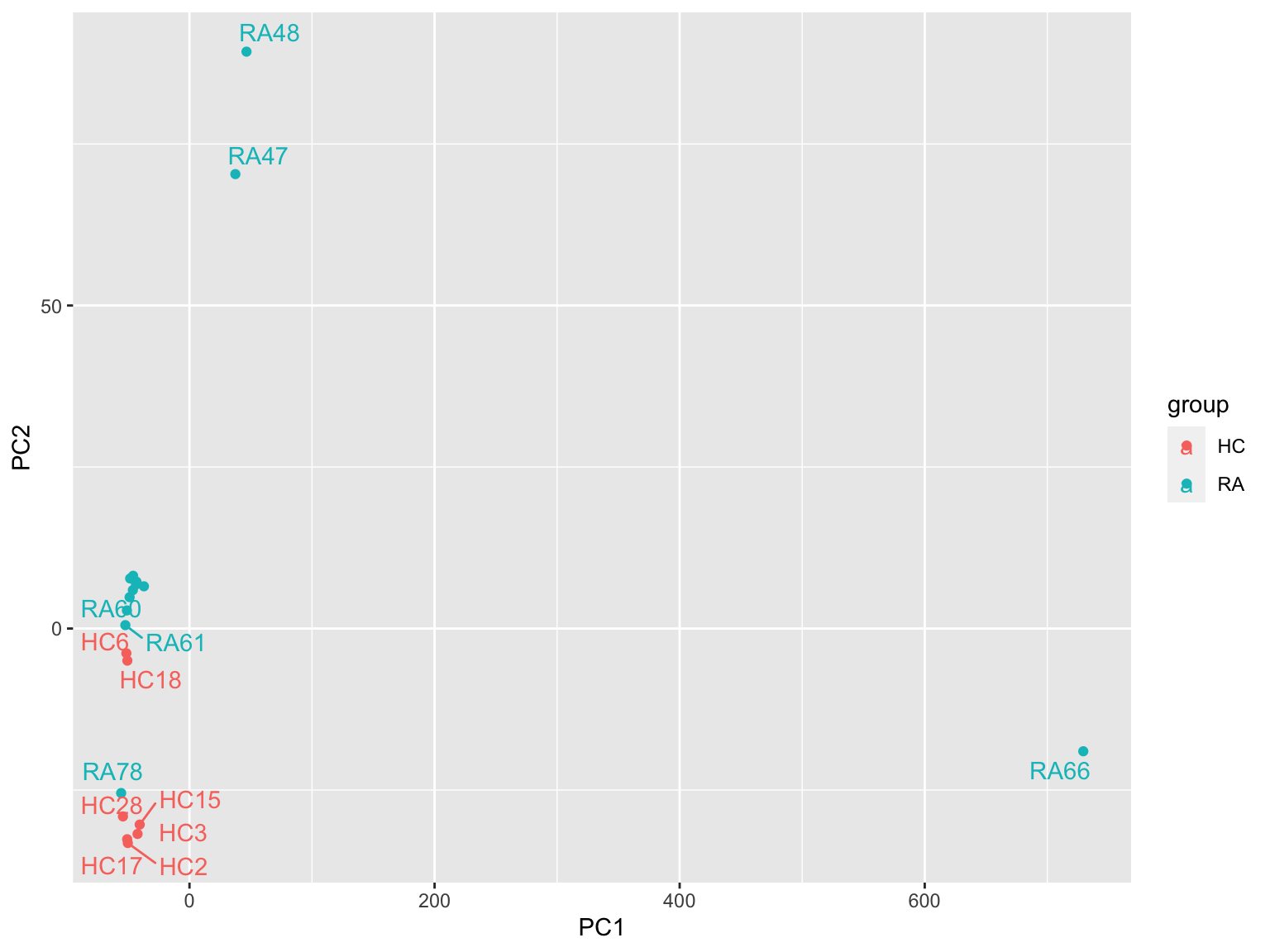 RA66, RA47 and RA48 looks like an outlier
RA66, RA47 and RA48 looks like an outlier
Remove three potential outliers and redo the preprocessing
Subset
diaSub <- seDIA[,! seDIA$sampleID %in% c("RA66","RA47","RA48")]
#remove proteins with more than 50% missing values
diaSub <- diaSub[rowSums(is.na(assay(diaSub)))/ncol(diaSub)<=0.5,]
dim(diaSub)[1] 5450 17Vst
protMat <- assay(diaSub)
fitVsn <- vsn::vsnMatrix(protMat)
normMat <- vsn::predict(fitVsn, newdata = protMat)
protNorm <- diaSub
assay(protNorm) <- normMatImputation
protImp <- DEP::impute(protNorm, "bpca")
assays(diaSub)[["norm"]] <- normMat
assays(diaSub)[["imputed"]] <- assay(protImp)Distribution after normalizaiton
countMat <- assays(diaSub)[["norm"]]
countTab <- countMat %>% as_tibble(rownames = "id") %>%
pivot_longer(-id) %>%
filter(!is.na(value))
ggplot(countTab, aes(x=name, y=value)) +
geom_boxplot() + geom_point()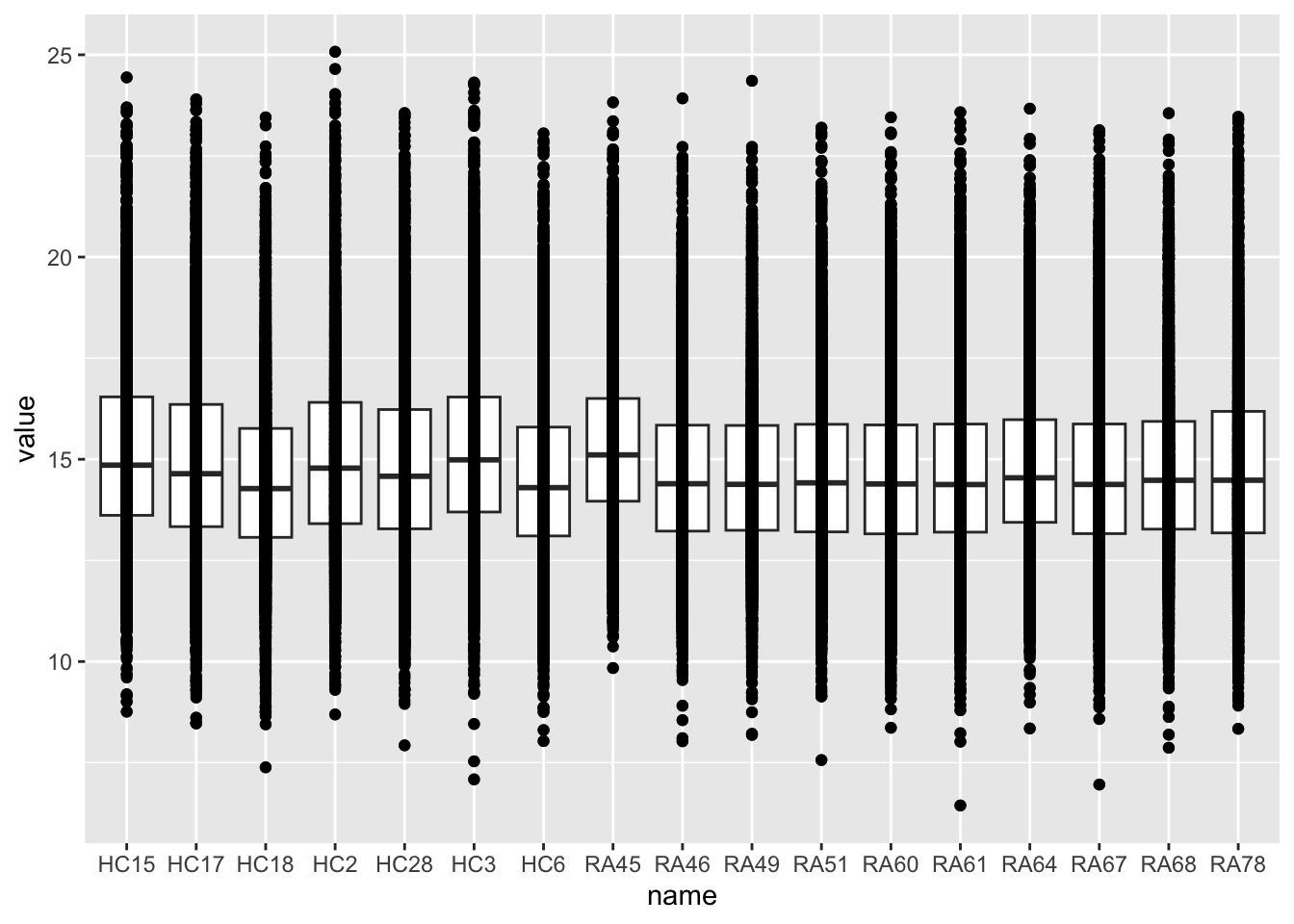
Redo pca
Color by group
plotMat <- assays(diaSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = group)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))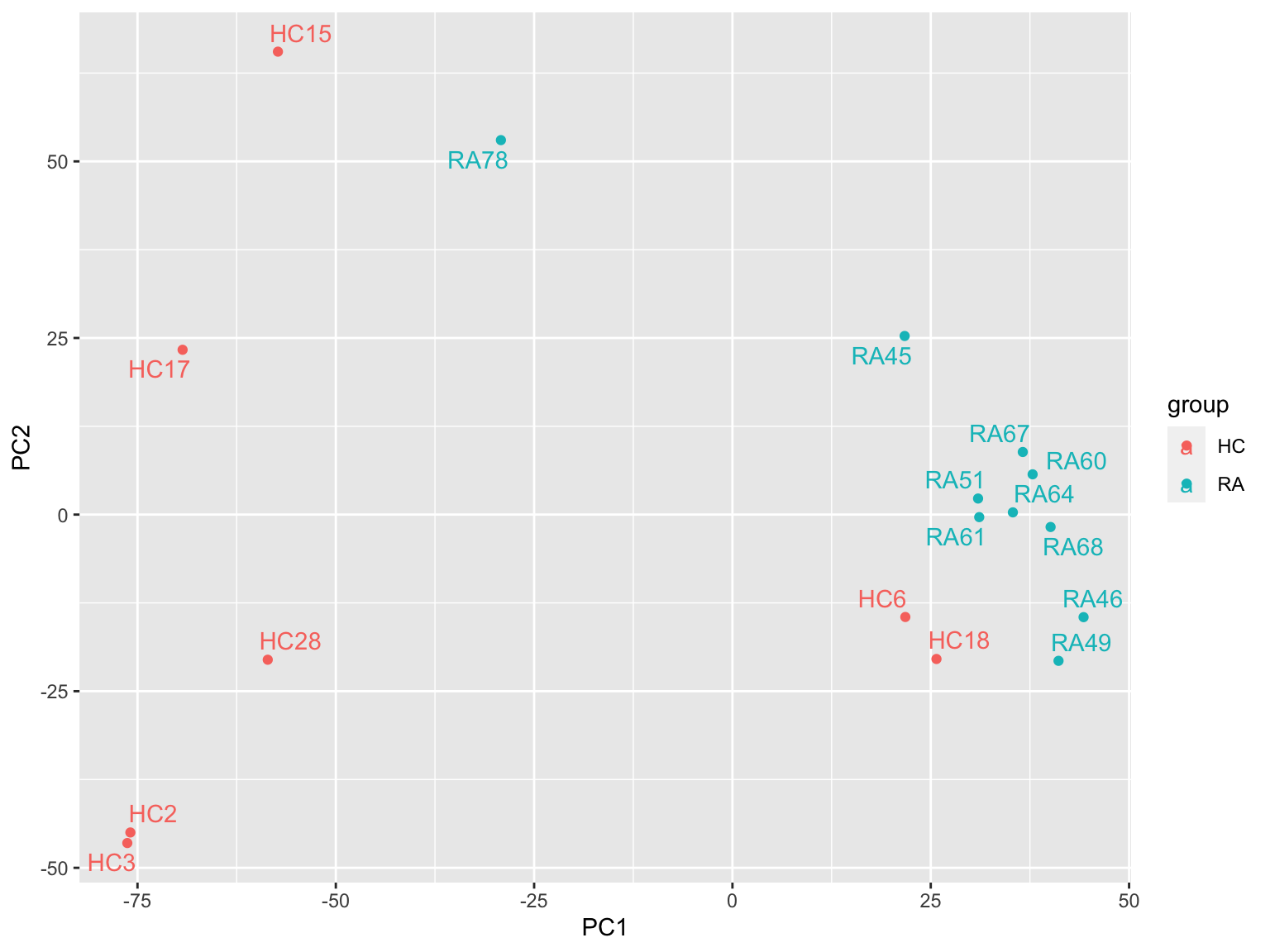 Looks better.
Looks better.
Color by quantStart
plotMat <- assays(diaSub)[["imputed"]]
prRes <- prcomp(t(plotMat), scale. = TRUE, center = TRUE)
plotTab <- prRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(as_tibble(colAnno, rownames = "sampleID"))
ggplot(plotTab, aes(x=PC1, y=PC2, col = quantStart)) +
geom_point() +
ggrepel::geom_text_repel(aes(label = sampleID))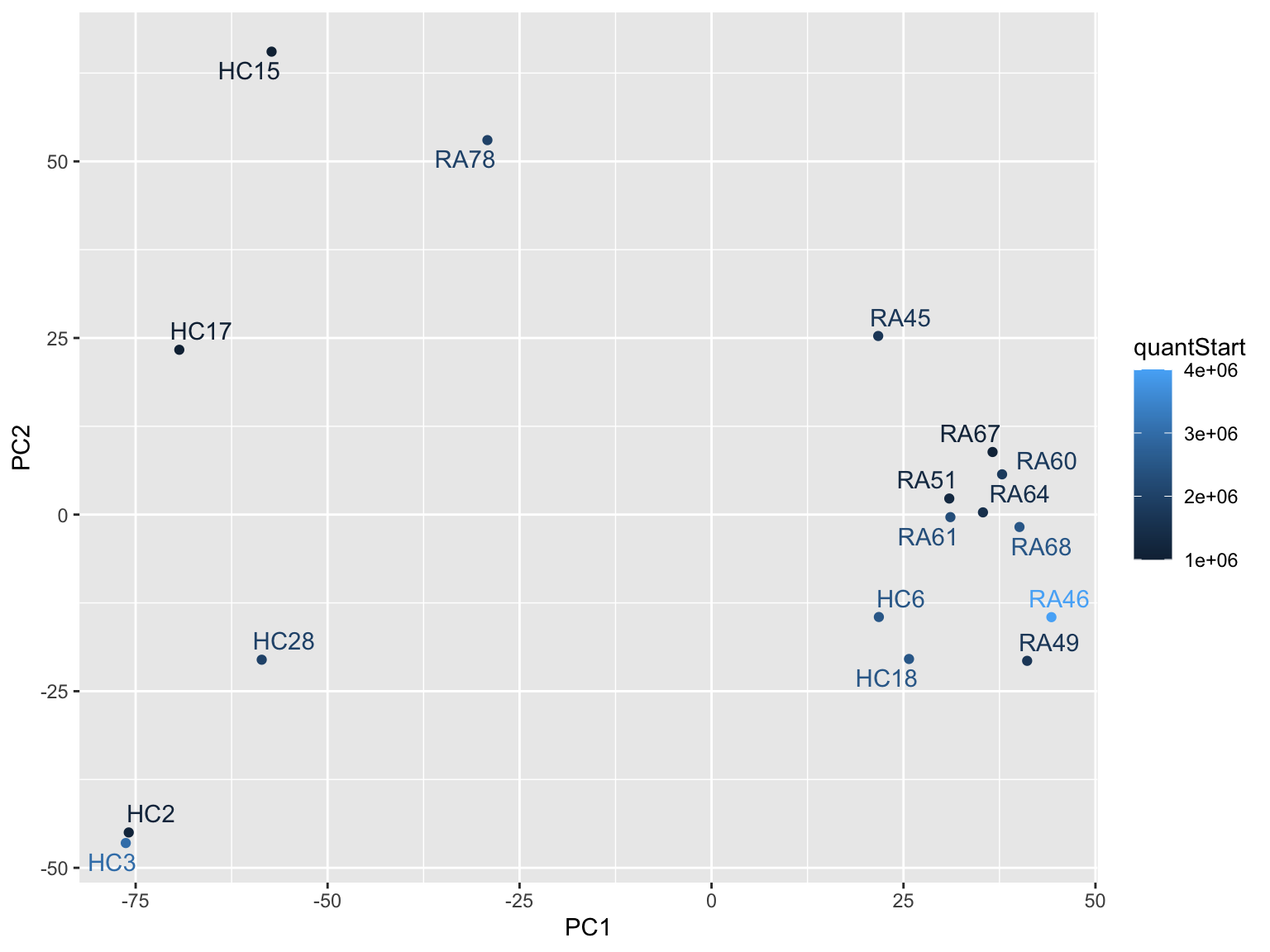
Assemble Multi-assay experiment object
library(MultiAssayExperiment)
maeObj <- MultiAssayExperiment(experiments = list(Metabolism = seMeta, Proteome = protSub, Proteome_DIA = diaSub,
Phosphoproteome = phosSub, PhosRatio = seRatio,
Methylation = methMat, FACS = seFacs),
colData = patTab %>% column_to_rownames("sampleID") %>% data.frame())
save(maeObj, file = "../output/maeObj.RData")
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] MultiAssayExperiment_1.22.0 pheatmap_1.0.12
[3] forcats_0.5.1 stringr_1.4.1
[5] dplyr_1.1.4.9000 purrr_0.3.4
[7] readr_2.1.2 tidyr_1.2.0
[9] tibble_3.2.1 ggplot2_3.4.1
[11] tidyverse_1.3.2 SummarizedExperiment_1.26.1
[13] GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[15] IRanges_2.30.0 S4Vectors_0.34.0
[17] MatrixGenerics_1.8.1 matrixStats_0.62.0
[19] jyluMisc_0.1.5 vsn_3.64.0
[21] Biobase_2.56.0 BiocGenerics_0.42.0
loaded via a namespace (and not attached):
[1] DEP_1.18.0 utf8_1.2.4 shinydashboard_0.7.2
[4] gmm_1.6-6 tidyselect_1.2.1 RSQLite_2.2.15
[7] AnnotationDbi_1.58.0 htmlwidgets_1.5.4 grid_4.2.0
[10] BiocParallel_1.30.3 norm_1.0-10.0 maxstat_0.7-25
[13] munsell_0.5.0 codetools_0.2-18 preprocessCore_1.58.0
[16] DT_0.23 withr_3.0.0 colorspace_2.0-3
[19] highr_0.9 knitr_1.39 rstudioapi_0.13
[22] ggsignif_0.6.3 mzID_1.34.0 labeling_0.4.2
[25] git2r_0.30.1 slam_0.1-50 GenomeInfoDbData_1.2.8
[28] KMsurv_0.1-5 bit64_4.0.5 farver_2.1.1
[31] rprojroot_2.0.3 vctrs_0.6.5 generics_0.1.3
[34] TH.data_1.1-1 xfun_0.31 sets_1.0-21
[37] R6_2.5.1 doParallel_1.0.17 clue_0.3-61
[40] MsCoreUtils_1.8.0 bitops_1.0-7 cachem_1.0.6
[43] fgsea_1.22.0 DelayedArray_0.22.0 assertthat_0.2.1
[46] promises_1.2.0.1 scales_1.2.0 multcomp_1.4-19
[49] googlesheets4_1.0.0 gtable_0.3.0 Cairo_1.6-0
[52] affy_1.74.0 sandwich_3.0-2 workflowr_1.7.0
[55] rlang_1.1.3 genefilter_1.78.0 mzR_2.30.0
[58] GlobalOptions_0.1.2 splines_4.2.0 rstatix_0.7.0
[61] gargle_1.2.0 impute_1.70.0 broom_1.0.0
[64] BiocManager_1.30.18 yaml_2.3.5 abind_1.4-5
[67] modelr_0.1.8 crosstalk_1.2.0 backports_1.4.1
[70] httpuv_1.6.6 tools_4.2.0 relations_0.6-12
[73] affyio_1.66.0 ellipsis_0.3.2 gplots_3.1.3
[76] jquerylib_0.1.4 RColorBrewer_1.1-3 MSnbase_2.22.0
[79] plyr_1.8.7 Rcpp_1.0.9 visNetwork_2.1.0
[82] zlibbioc_1.42.0 RCurl_1.98-1.7 ggpubr_0.4.0
[85] GetoptLong_1.0.5 cowplot_1.1.1 zoo_1.8-10
[88] ggrepel_0.9.1 haven_2.5.0 cluster_2.1.3
[91] exactRankTests_0.8-35 fs_1.5.2 magrittr_2.0.3
[94] magick_2.7.3 data.table_1.14.8 circlize_0.4.15
[97] reprex_2.0.1 survminer_0.4.9 pcaMethods_1.88.0
[100] googledrive_2.0.0 mvtnorm_1.1-3 ProtGenerics_1.28.0
[103] hms_1.1.1 shinyjs_2.1.0 mime_0.12
[106] evaluate_0.15 xtable_1.8-4 XML_3.99-0.10
[109] readxl_1.4.0 gridExtra_2.3 shape_1.4.6
[112] compiler_4.2.0 KernSmooth_2.23-20 ncdf4_1.19
[115] crayon_1.5.2 htmltools_0.5.4 later_1.3.0
[118] tzdb_0.3.0 lubridate_1.8.0 DBI_1.1.3
[121] dbplyr_2.2.1 ComplexHeatmap_2.12.1 tmvtnorm_1.5
[124] MASS_7.3-58 Matrix_1.5-4 car_3.1-0
[127] cli_3.6.2 imputeLCMD_2.1 marray_1.74.0
[130] parallel_4.2.0 igraph_1.3.4 pkgconfig_2.0.3
[133] km.ci_0.5-6 piano_2.12.0 MALDIquant_1.21
[136] xml2_1.3.3 foreach_1.5.2 annotate_1.74.0
[139] bslib_0.4.1 XVector_0.36.0 drc_3.0-1
[142] rvest_1.0.2 digest_0.6.30 Biostrings_2.64.0
[145] rmarkdown_2.14 cellranger_1.1.0 fastmatch_1.1-3
[148] survMisc_0.5.6 shiny_1.7.4 gtools_3.9.3
[151] rjson_0.2.21 lifecycle_1.0.4 jsonlite_1.8.3
[154] carData_3.0-5 limma_3.52.2 fansi_1.0.6
[157] pillar_1.9.0 lattice_0.20-45 KEGGREST_1.36.3
[160] fastmap_1.1.0 httr_1.4.3 plotrix_3.8-2
[163] survival_3.4-0 glue_1.7.0 png_0.1-7
[166] iterators_1.0.14 bit_4.0.4 stringi_1.7.8
[169] sass_0.4.2 blob_1.2.3 memoise_2.0.1
[172] caTools_1.18.2