Compare the signature of Doxorubicin resistant and sensitive DLBCL cell lines (using continous response variable)
Junyan Lu
2022-06-10
Last updated: 2022-07-08
Checks: 4 2
Knit directory: combiDLBCL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220425) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
- unnamed-chunk-80
- unnamed-chunk-84
- unnamed-chunk-88
- unnamed-chunk-92
- unnamed-chunk-96
To ensure reproducibility of the results, delete the cache directory
Doxo_resistant_cache and re-run the analysis. To have
workflowr automatically delete the cache directory prior to building the
file, set delete_cache = TRUE when running
wflow_build() or wflow_publish().
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load libraries and datasets
Data preprocessing
Drug screen data (the cell line screen data from Tobias)
Read data
load("../data/Screen.CL19.RData")
screenData <- filter(Screen.CL19, str_detect(Entity, "DLBCL"), Drug == "Doxorubicine", TimePoint == "48 h") %>%
mutate(Subtype = str_remove(Entity,"DLBCL ")) %>%
select(Name, Drug.Conc, Duplicate, Normalized, Subtype)Using 48h Doxorubicin treatment response as the continous response variable
Some cell lines have duplicates, check the reproducibility
dupCell <- unique(filter(screenData, Duplicate == "2")$Name)
compareTab <- filter(screenData, Name %in% dupCell) %>%
mutate(Duplicate = paste0("R",Duplicate)) %>%
pivot_wider(names_from = Duplicate, values_from = Normalized)
ggplot(compareTab, aes(x=R1, y=R2, col = factor(Drug.Conc))) +
geom_point() +
geom_abline(intercept = 0, slope = 1, col = "red") +
facet_grid(~Name)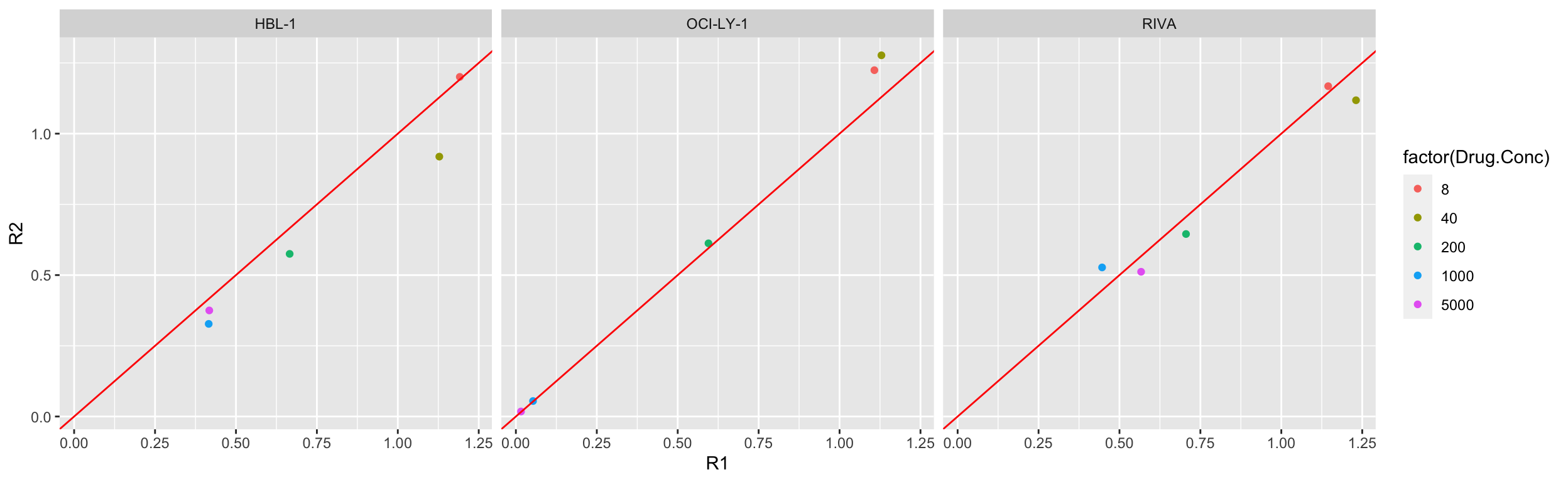 Look fine, the duplicates can be averaged.
Look fine, the duplicates can be averaged.
screenData <- group_by(screenData, Name, Drug.Conc, Subtype) %>%
summarise(normVal = mean(Normalized)) %>%
ungroup()Dose response of Doxorubicin in DLBCL cell lines
plotTab <- screenData
p <- ggplot(plotTab, aes(x=Drug.Conc, y=normVal, group = Name, col = Name)) +
geom_line() + geom_point() +
scale_x_log10() + theme_bw() +
coord_cartesian(ylim = c(0,1.5)) +
#ggtitle(paste0(rec$Drug_B)) +
theme(legend.position = "right")
p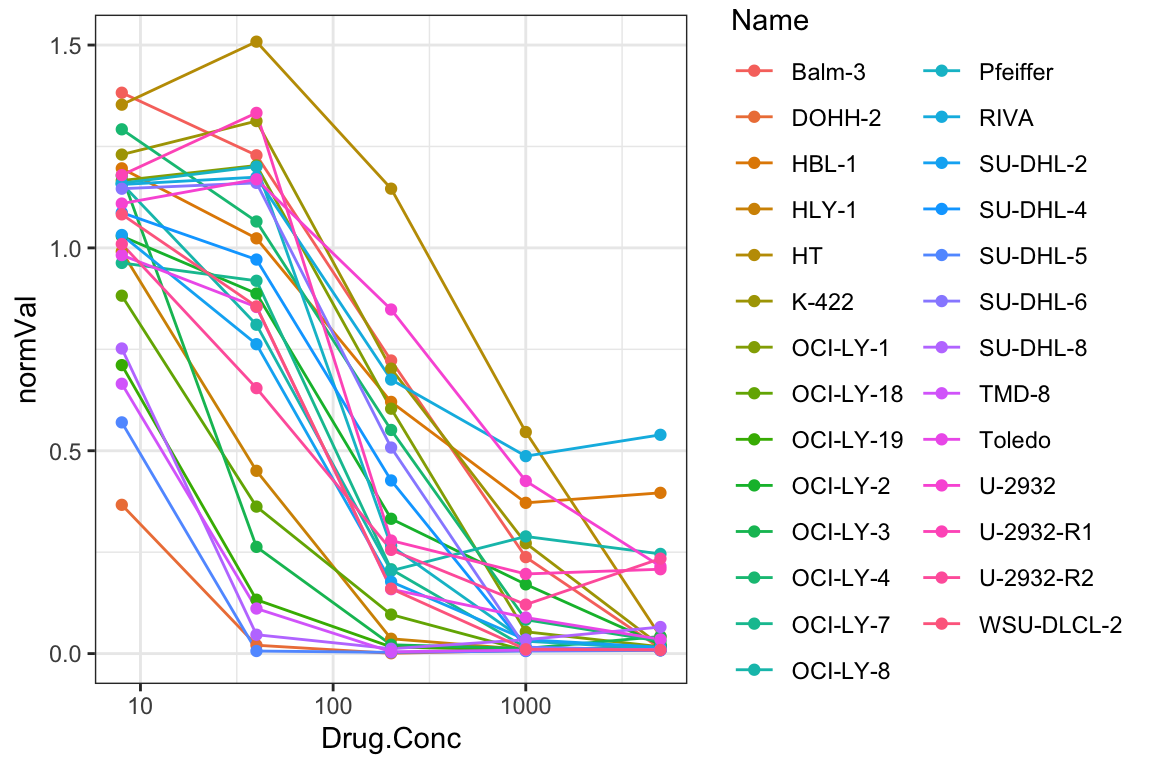 It’s difficult to define resistant and sensitive groups. Better to use
continues response based on AUC
It’s difficult to define resistant and sensitive groups. Better to use
continues response based on AUC
aucTab <- screenData %>% group_by(Name,Subtype) %>%
summarise(auc = calcAUC(normVal, Drug.Conc)) %>%
ungroup() %>%
arrange(auc) %>%
mutate(Name = factor(Name, levels = Name))
ggplot(aucTab, aes(x=Name, y=auc, col = Subtype)) +
geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))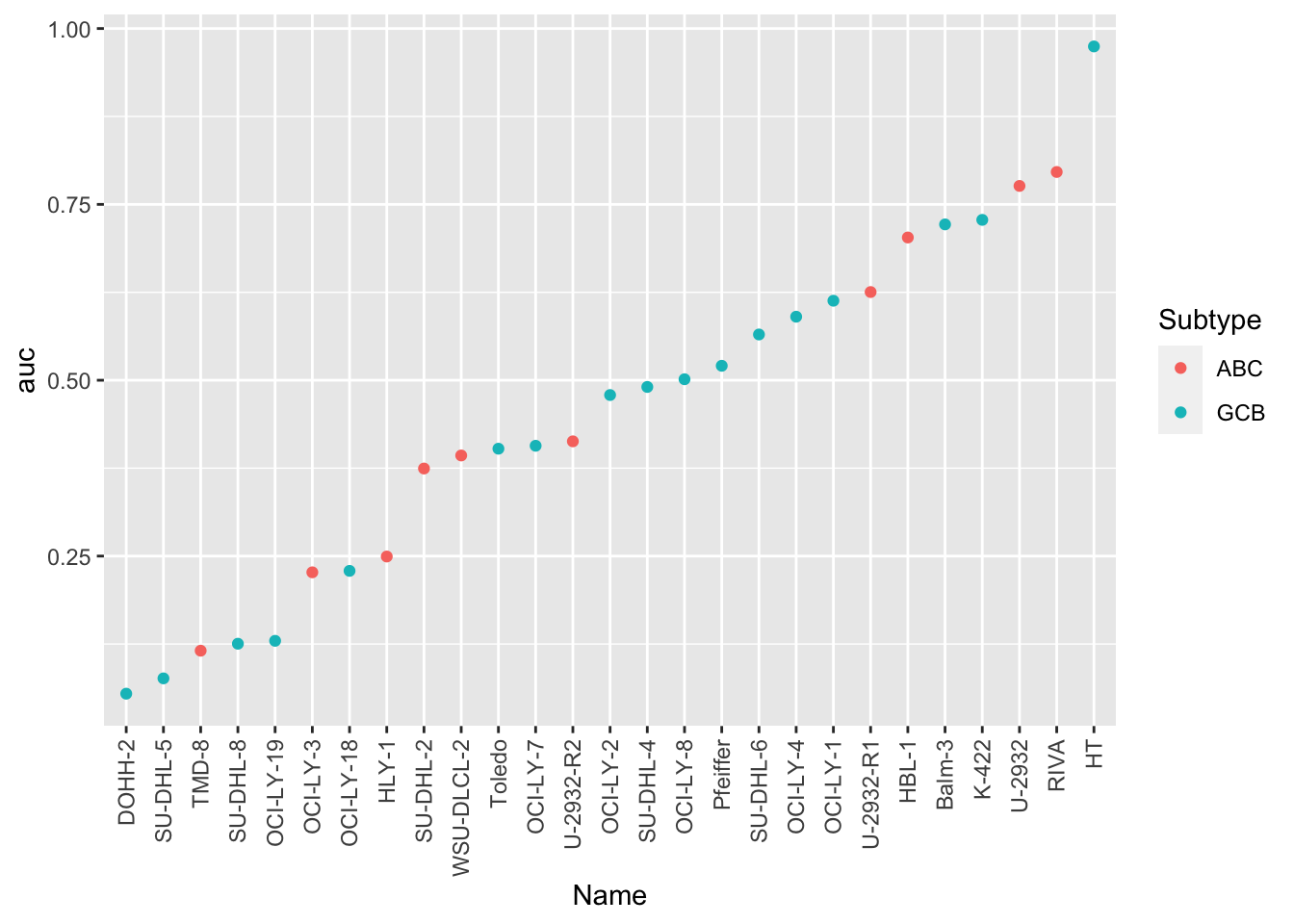
Whether Doxorubicine response is associated with ABC/GCB subtype?
ggplot(aucTab, aes(x=Subtype, y = auc)) +
geom_boxplot() + geom_point()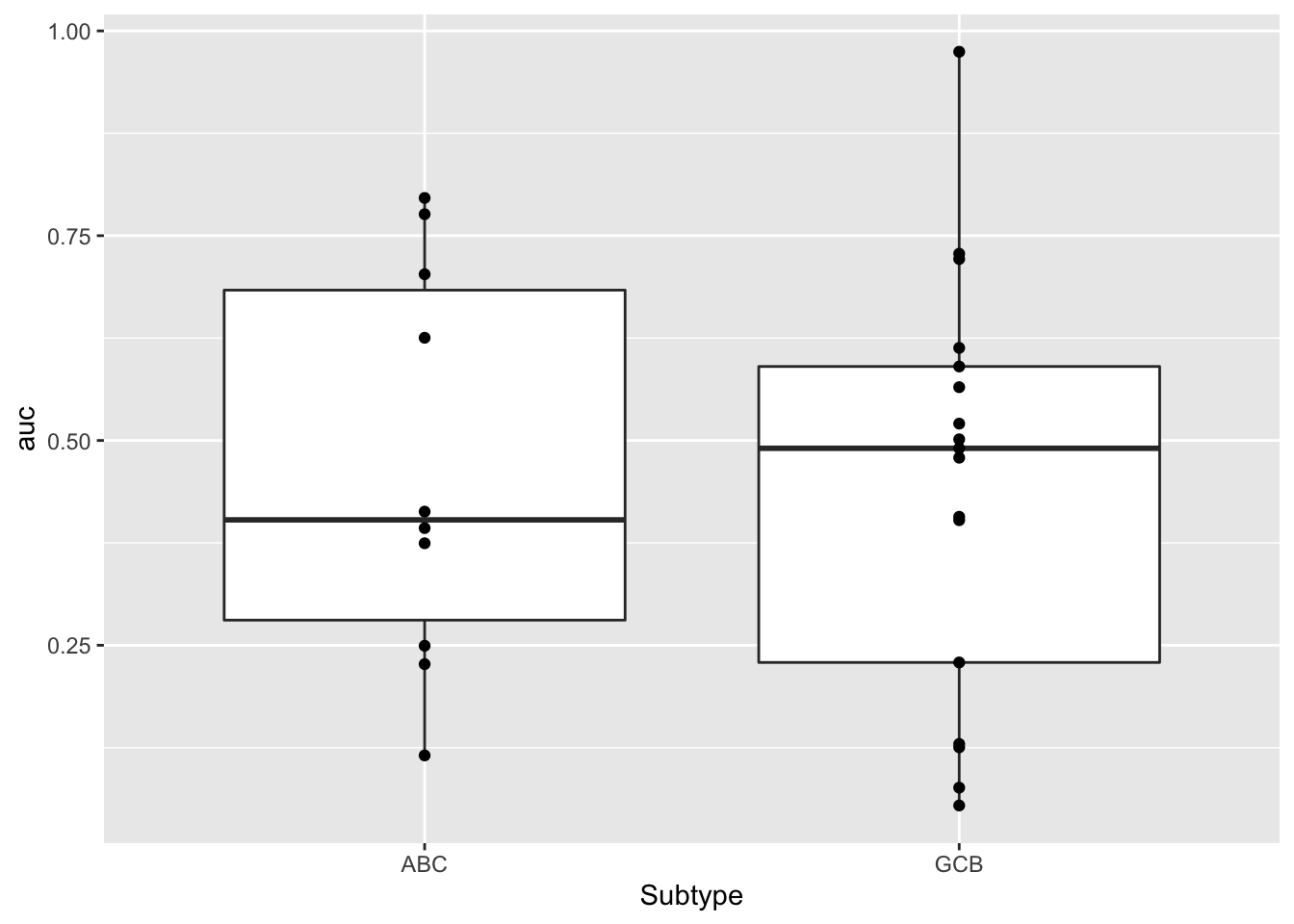
t.test(auc~Subtype, aucTab)
Welch Two Sample t-test
data: auc by Subtype
t = 0.20033, df = 19.886, p-value = 0.8433
alternative hypothesis: true difference in means between group ABC and group GCB is not equal to 0
95 percent confidence interval:
-0.1863762 0.2259621
sample estimates:
mean in group ABC mean in group GCB
0.4673122 0.4475192 No
Genomics
Processing genomics data
Load genomics
load("../data/SVs_filtered.RData")
svTab <- filter(svTab, Name %in% aucTab$Name)Summarise mutations: count as gene mutation if there is at least one mutation within gene
mutTab <- group_by(svTab, Name, Gene) %>% summarise(n = length(Name)) %>%
arrange(desc(n))
#Get mutations occured at least in three cell lines
geneCount <- group_by(mutTab, Gene) %>% summarise(n=length(Name)) %>%
filter(n>=3) %>% arrange(desc(n)) %>%
mutate(Gene = factor(Gene, levels = Gene))There are too many mutations. Manual curation maybe needed.
Occurrence of mutations among cell lines, only mutations occurred at least 3 times are considered
ggplot(geneCount, aes(x=Gene, y=n)) +
geom_bar(stat = "identity") +
theme_bw() + theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))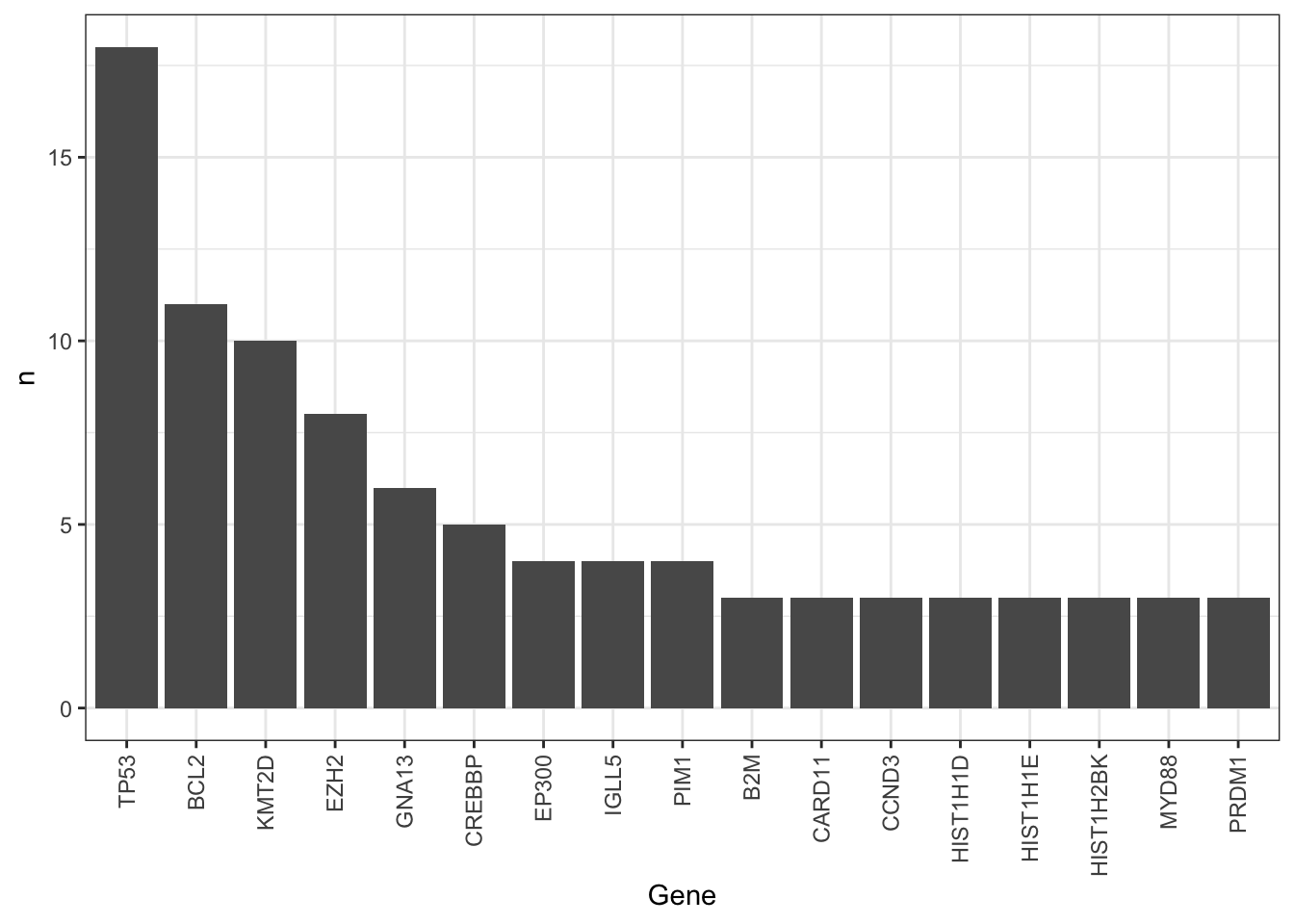
T-test
geneTab <- filter(mutTab, Gene %in% geneCount$Gene) %>%
mutate(status =1) %>% select(Name, Gene, status) %>%
pivot_wider(names_from = "Gene", values_from = "status") %>%
mutate_all(replace_na,0) %>%
pivot_longer(-Name, names_to = "Gene", values_to = "status") %>%
ungroup()
testTab <- left_join(geneTab, aucTab, by = "Name")
geneMat <- geneTab %>% pivot_wider(names_from = Name, values_from = status) %>%
column_to_rownames("Gene") %>% as.matrix()
omicList <- list(gene = geneMat)
resTab <- group_by(testTab, Gene) %>% nest() %>%
mutate(m = map(data, ~t.test(auc~status,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Gene, p.value) %>%
arrange(p.value)
head(resTab)# A tibble: 6 × 2
# Groups: Gene [6]
Gene p.value
<chr> <dbl>
1 TP53 0.000279
2 EZH2 0.0510
3 PRDM1 0.0529
4 KMT2D 0.114
5 PIM1 0.237
6 HIST1H1E 0.353 TP53 is the most significant one
Dose response of Doxorubicine with TP53 annotated
plotTab <- screenData %>%
left_join(filter(geneTab, Gene == "TP53"), by = "Name") %>%
mutate(TP53 = case_when(
status == 0 ~ "WT",
status == 1 ~ "Mut"
))
p <- ggplot(plotTab, aes(x=Drug.Conc, y=normVal,
group = Name, col = factor(TP53))) +
geom_line() + geom_point() +
scale_x_log10() + theme_bw() +
coord_cartesian(ylim = c(0,1.5)) +
theme(legend.position = "right")
p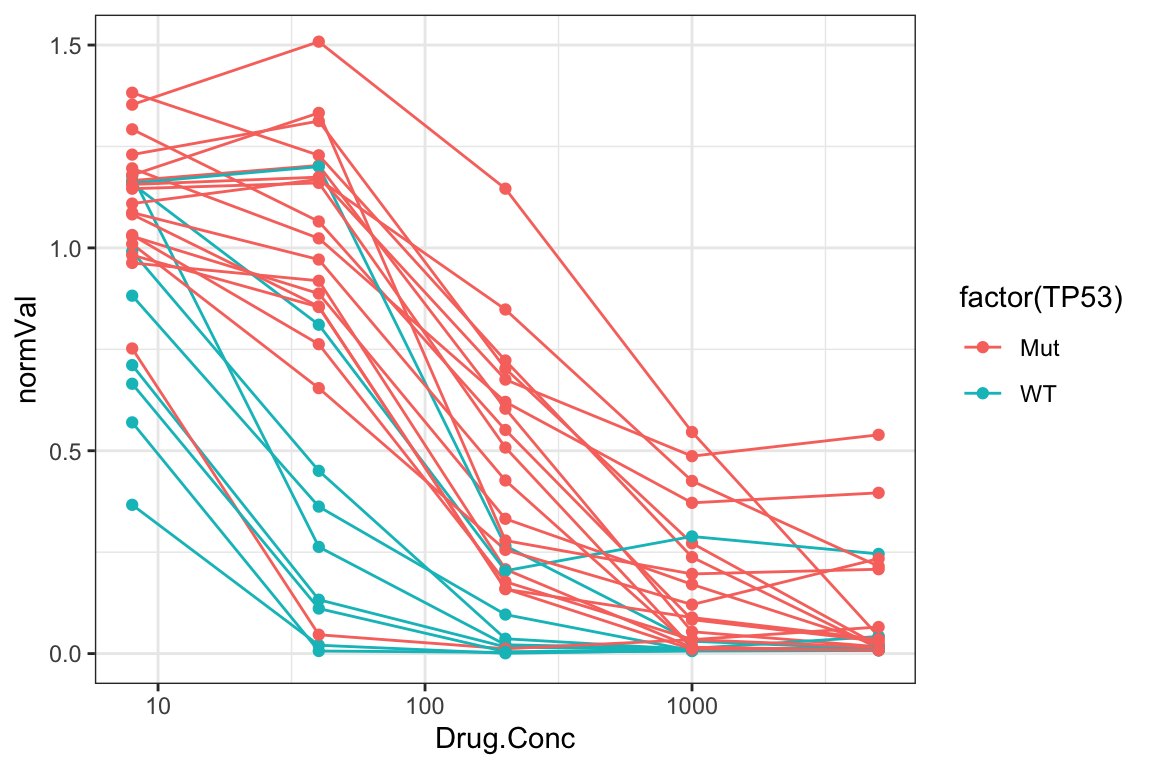
Add TP53 status to the AUC table
aucTab <- aucTab %>%
left_join(filter(geneTab, Gene == "TP53"), by = "Name") %>%
mutate(TP53 = case_when(
status == 0 ~ "WT",
status == 1 ~ "Mut"
)) %>%
select(-Gene, -status) %>%
arrange(auc)
plotTab <- aucTab %>% mutate(Name = factor(Name, levels = Name))
ggplot(plotTab, aes(x=Name,y=auc,col=TP53)) +
geom_point() +
theme(axis.text.x = element_text(angle = 90, hjust =1, vjust = 0.5))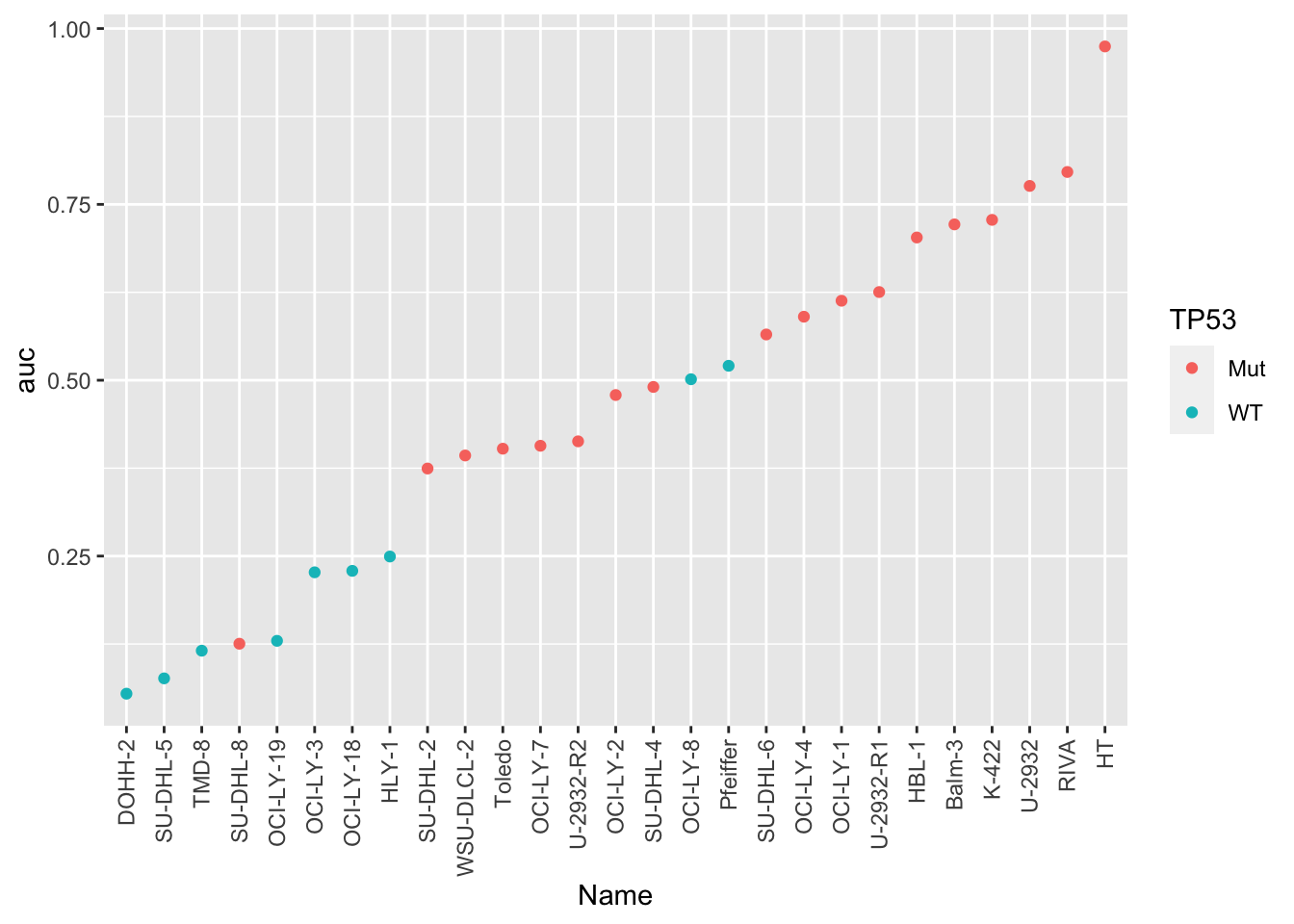 The response is mostly accord with TP53 mutation status. Only a few cell
lines are outliers:
The response is mostly accord with TP53 mutation status. Only a few cell
lines are outliers:
SU-DHL-8 is TP53 mutated, but also sensitive.
OCI-LY-8 and Pfeffer are WT but rather resistant to Doxorubicine
Association with proteomics
Preprocessing proteomic data
Data distribution
load("../data/ProtWide.RData")
ProtWide <- ProtWide[,colnames(ProtWide) %in% aucTab$Name]
protMat <- ProtWide
dim(ProtWide)[1] 4873 27Median normalization (not performed)
#protMatNorm <- PhosR::medianScaling(protMat, scale = FALSE)
protMatNorm <- protMat
boxplot(protMatNorm)
#protNorm <- protData
#assay(protNorm) <- protMatNormCreate assay experiment object
protTab <- protMatNorm %>% as_tibble(rownames = "uniprotID") %>%
pivot_longer(-uniprotID, names_to = "Name", values_to = "count") %>%
left_join(aucTab, by = "Name") %>%
mutate(symbol = uniprotID, cellLine = Name) %>%
filter(!symbol %in% c("",NA), !is.na(auc))
protSub <- jyluMisc::tidyToSum(protTab, rowID = "uniprotID",colID = "Name",
values = "count", annoRow = "symbol", annoCol = c("auc","TP53","Subtype","cellLine"))Identify proteins differentially expressed
Prefilter proteins that did not change
sds <- genefilter::rowSds(assay(protSub))
protSub <- protSub[sds > genefilter::shorth(sds),]
dim(protSub)[1] 3268 27Differential protein expression using proDA
protMat <- assay(protSub)
omicList[["protein"]] <- protMat
fit <- proDA(protMat, design = ~ auc,
col_data = colData(protSub))
resTab <- test_diff(fit, contrast = "auc") %>%
arrange(pval) %>%
mutate(symbol = rowData(protSub[name,])$symbol)
resTab.base.embl <- resTabhist(resTab$pval) Stronger associations can be observed
Stronger associations can be observed
Proteins with p-value < 0.05
resTab.sig <- filter(resTab, pval < 0.05)
resTab.sig %>% select(symbol, pval, adj_pval, diff) %>%
mutate_if(is.numeric, formatC, digits=1) %>%
DT::datatable()Plot top 9 examples
pList <- lapply(seq(9), function(i) {
rec <- resTab.sig[i,]
plotTab <- tibble(expr = protMat[rec$name,],
auc = protSub$auc,
TP53 = protSub$TP53,
Name = colnames(protSub))
ggplot(plotTab, aes(x=auc, y=expr)) +
geom_point(aes(col = TP53)) +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(rec$symbol) +
geom_smooth(method="lm", se= FALSE) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList,ncol=3)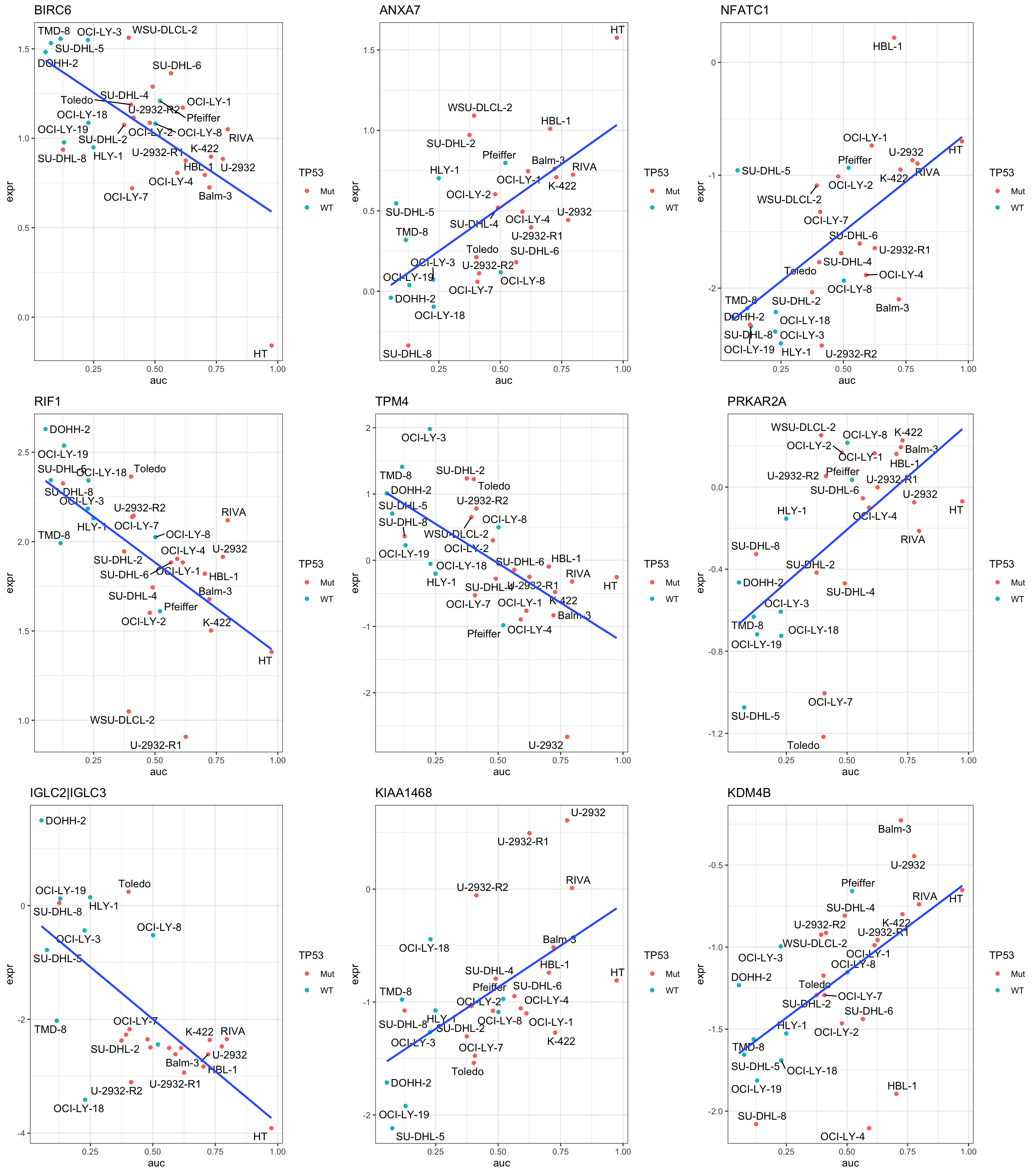
Enrichment analysis
gmts = list(H= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt",
C6 = "../data/gmts/c6.all.v6.2.symbols.gmt")
inputTab <- resTab %>% filter(pval < 0.1) %>%
distinct(symbol, .keep_all = TRUE) %>%
select(symbol, t_statistic) %>% data.frame() %>% column_to_rownames("symbol")
enRes <- list()
enRes[["Hallmark"]] <- runGSEA(inputTab, gmts$H, "page")
enRes[["KEGG"]] <- runGSEA(inputTab, gmts$KEGG,"page")
enRes[["Perturbation"]] <- runGSEA(inputTab, gmts$C6,"page")
p <- plotEnrichmentBar(enRes, pCut =0.05, ifFDR= FALSE)
cowplot::plot_grid(p)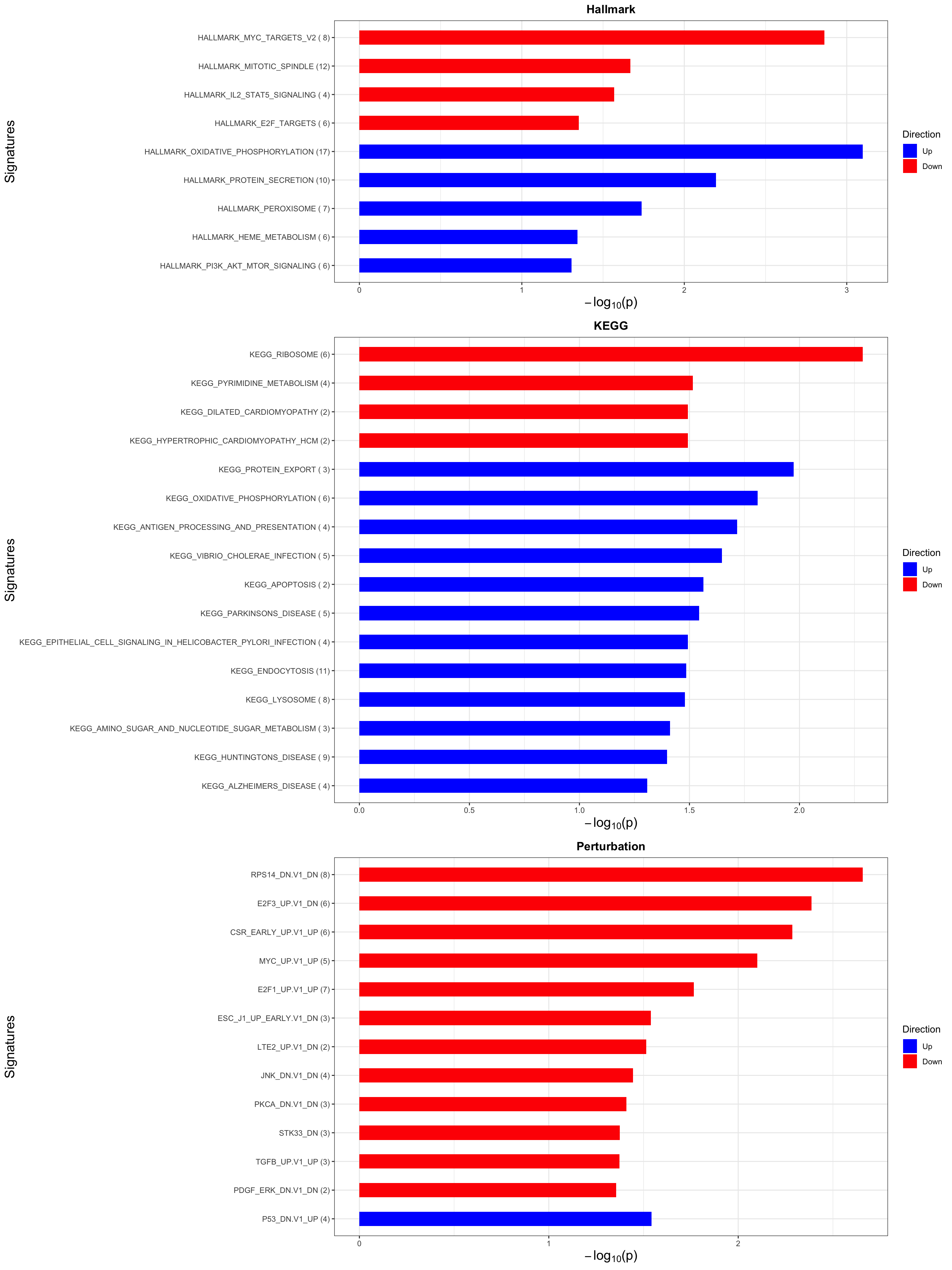
Focus on proteins from Fatty acid metabolism pathway
geneList <- piano::loadGSC(gmts$H)$gsc$HALLMARK_FATTY_ACID_METABOLISM
plotGene <- filter(filter(resTab, pval <= 0.05), symbol%in% geneList )
pList <- lapply(seq(nrow(plotGene)), function(i) {
rec <- plotGene[i,]
plotTab <- tibble(expr = protMat[rec$name,],
auc = protSub$auc,
TP53 = protSub$TP53,
Name = colnames(protSub))
ggplot(plotTab, aes(x=auc, y=expr)) +
geom_point(aes(col = TP53)) +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(rec$symbol) +
geom_smooth(method="lm", se= FALSE) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList,ncol=3)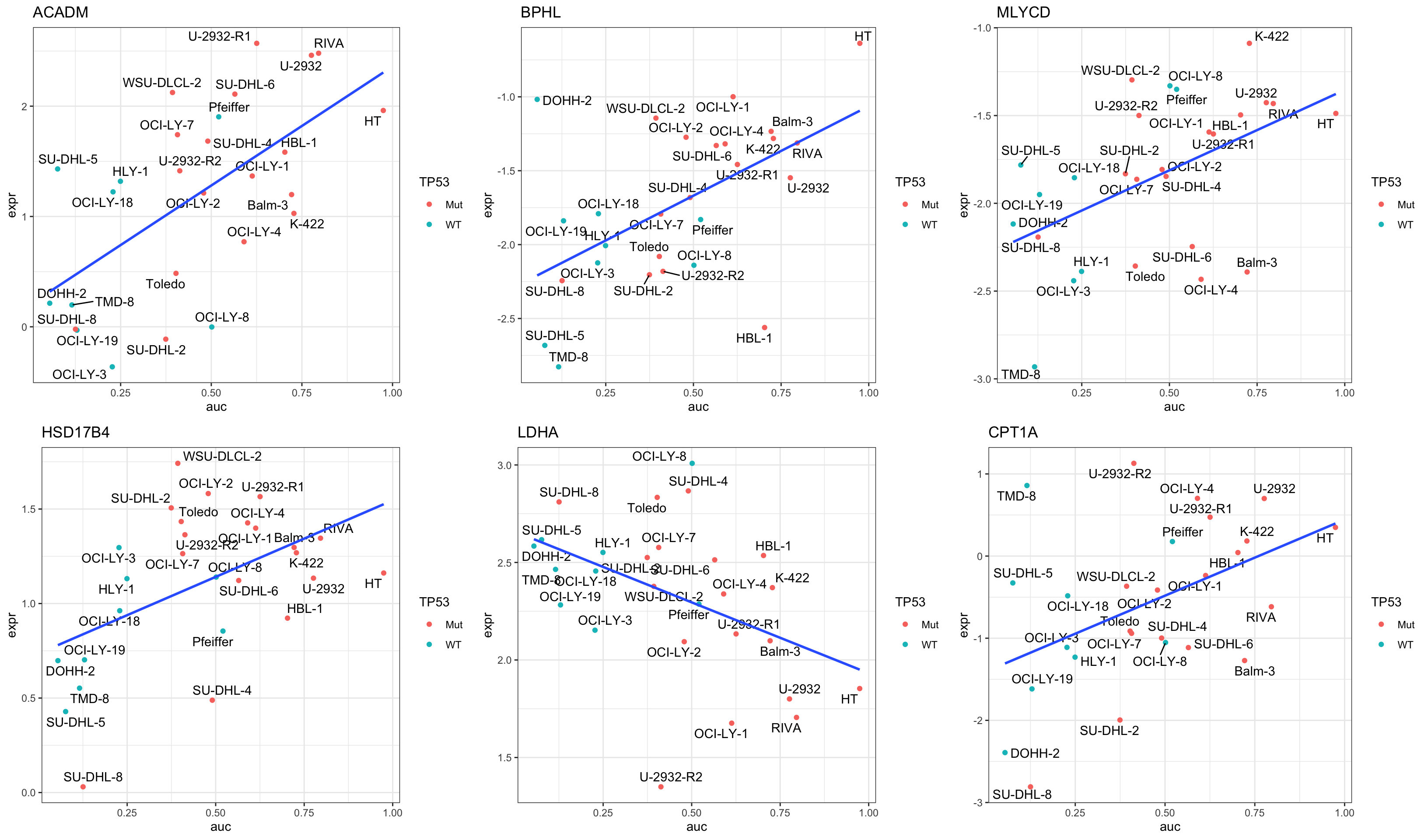
Metabolimics
Process metabolomic data
Normalization (not performed)
metaData <- readRDS("../data/SC005_SummarizedExperiment_metabolomics.RDS")
metaData <- metaData[,metaData$cell.line %in% aucTab$Name & metaData$condition =="U"]
metaMat <- assay(metaData)
boxplot(metaMat)
#metaMatNorm <- PhosR::medianScaling(metaMat, scale = FALSE)
metaMatNorm <- metaMat
boxplot(metaMatNorm)
metaNorm <- metaData
assay(metaNorm) <- metaMatNorm
assayNames(metaNorm) <- "norm"Average technical replicates for each cell line
metaTab <- jyluMisc::sumToTidy(metaNorm) %>%
group_by(cell.line, rowID, metabolite, class, condition) %>%
summarise(count = mean(norm, na.rm=TRUE)) %>%
dplyr::rename(symbol = metabolite, cellLine = cell.line) %>%
mutate(colID = paste0(cellLine,"_", condition)) %>%
ungroup()
metaSub <- jyluMisc::tidyToSum(metaTab, rowID = "rowID", colID = "colID",
values = "count", annoRow = "symbol",
annoCol = c("condition", "cellLine"))
#add additional annotations
metaSub$auc <- aucTab[match(metaSub$cellLine, aucTab$Name),]$auc
metaSub$TP53 <- aucTab[match(metaSub$cellLine, aucTab$Name),]$TP53
metaSub$Subtype <- aucTab[match(metaSub$cellLine, aucTab$Name),]$Subtype
#remove uncessary samples and records
metaSub <- metaSub[!rowData(metaSub)$symbol %in% c("",NA), !is.na(metaSub$auc)]
dim(metaSub)[1] 286 12colData(metaSub) %>% data.frame() %>% DT::datatable()Differential metabolites abundance
Differential metabolites abundance
metaMat <- assay(metaSub)
omicList[["metabolite"]] <- metaMat
designMat <- model.matrix(~ metaSub$auc)
fit <- lmFit(metaMat, designMat)
fit2 <- eBayes(fit)
resTab <- topTable(fit2, number= Inf, coef= "metaSub$auc") %>%
dplyr::rename(pval = P.Value, adj_pval = adj.P.Val) %>%
arrange(pval) %>%
as_tibble(rownames = "name") %>%
mutate(symbol = rowData(metaSub[name,])$symbol)hist(resTab$pval) Not strong difference
Not strong difference
metabolites with p-value < 0.05
resTab.sig <- filter(resTab, pval < 0.05)
resTab.sig %>% select(symbol, pval, adj_pval, logFC, t) %>%
mutate_if(is.numeric, formatC, digits=1) %>%
DT::datatable()Plot top 9 examples
pList <- lapply(seq(9), function(i) {
rec <- resTab.sig[i,]
plotTab <- tibble(expr = metaMat[rec$name,],
auc = metaSub$auc,
TP53 = metaSub$TP53,
Name = colnames(metaSub))
ggplot(plotTab, aes(x=auc, y=expr)) +
geom_point(aes(col = TP53)) +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(rec$symbol) +
geom_smooth(method="lm", se= FALSE) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList,ncol=3)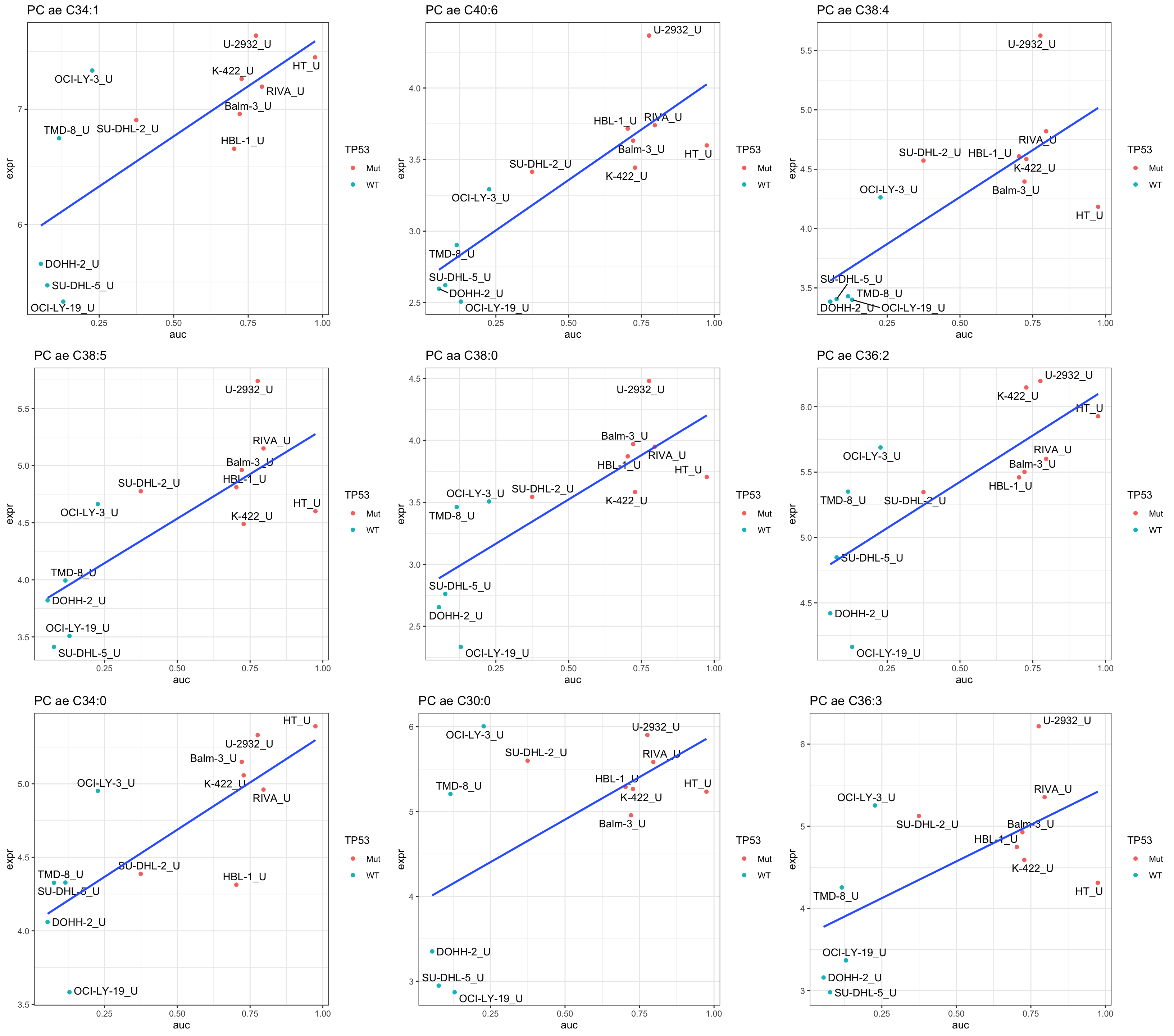
Differential gene expression analysis based on public data
Proprocessing
load("../data/DepMap_GEXwide.RData")
exprMat <- t(DepMap_GEXwide)
exprMat <- exprMat[,colnames(exprMat) %in% aucTab$Name]
# Remove low count genes
exprMat <- exprMat[rowMedians(exprMat) >0,]
dim(exprMat)[1] 15654 14boxplot(exprMat)
vstObj <- vsn::vsnMatrix(exprMat)
#exprMat <- vsn::predict(vstObj, exprMat)
#boxplot(exprMat)
#save process data
#save(exprMat, file = "gene_exprMat.RData")Invariance filtering
sds <- genefilter::rowSds(exprMat)
exprMat <- exprMat[sds > genefilter::shorth(sds),]
dim(exprMat)[1] 8392 14omicList[["rna"]] <- exprMatDifferential expression using limma
colTab <- aucTab[match(colnames(exprMat), aucTab$Name),] %>%
column_to_rownames("Name") %>% data.frame()
designMat <- model.matrix(~auc, colTab)
fit <- lmFit(exprMat, designMat)
fit2 <- eBayes(fit)
resTab <- topTable(fit2, number = Inf, coef = "auc") %>%
as_tibble(rownames = "symbol")
hist(resTab$P.Value)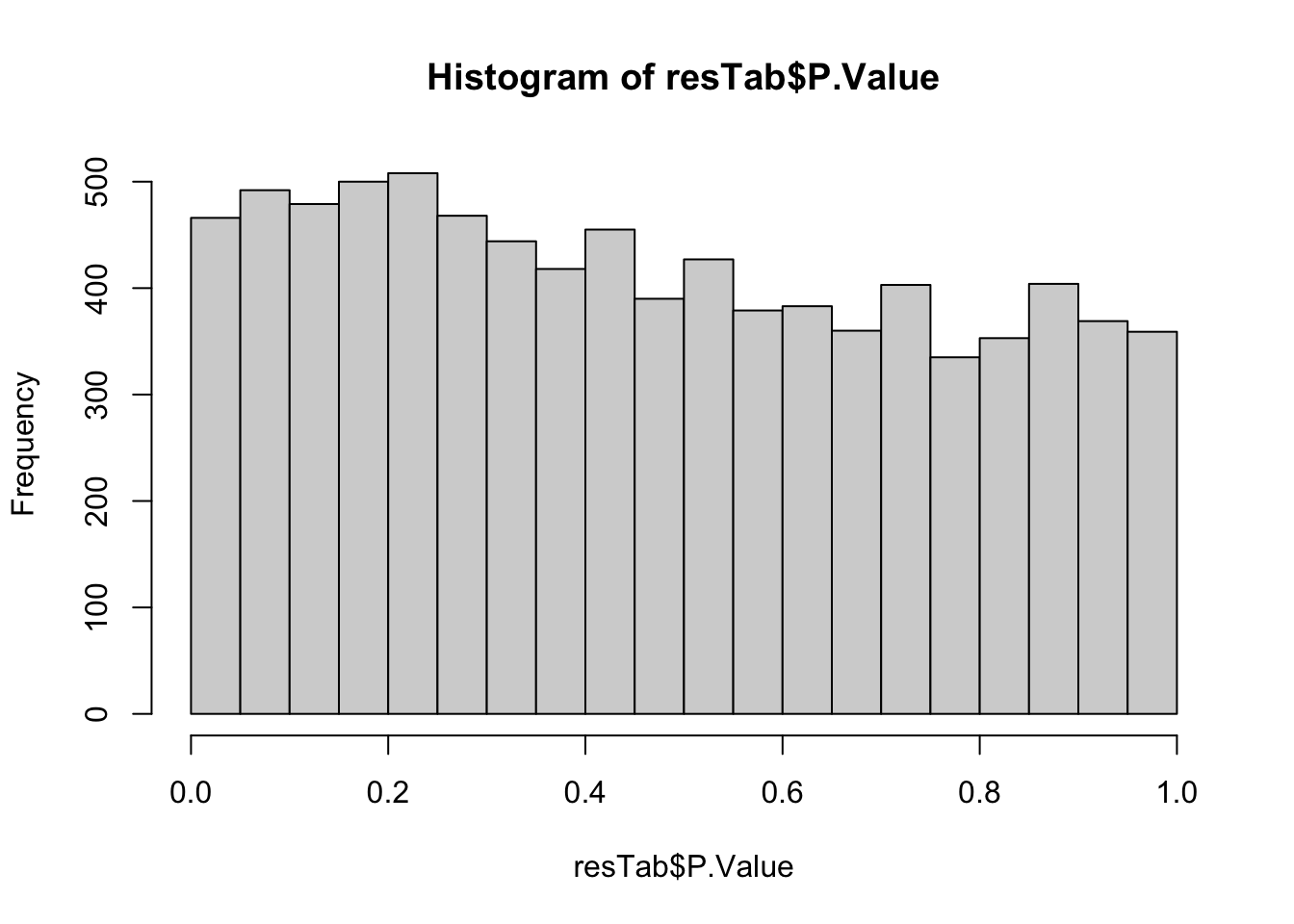
Associations with p <= 0.05
resTab.sig <- filter(resTab, P.Value <= 0.05)
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2) %>% DT::datatable()Plot top 9 examples
pList <- lapply(seq(9), function(i) {
rec <- resTab.sig[i,]
plotTab <- tibble(expr = exprMat[rec$symbol,],
auc = colTab$auc,
TP53 = colTab$TP53,
Name = colnames(exprMat))
ggplot(plotTab, aes(x=auc, y=expr)) +
geom_point(aes(col = TP53)) +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(rec$symbol) +
geom_smooth(method="lm", se= FALSE) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList,ncol=3)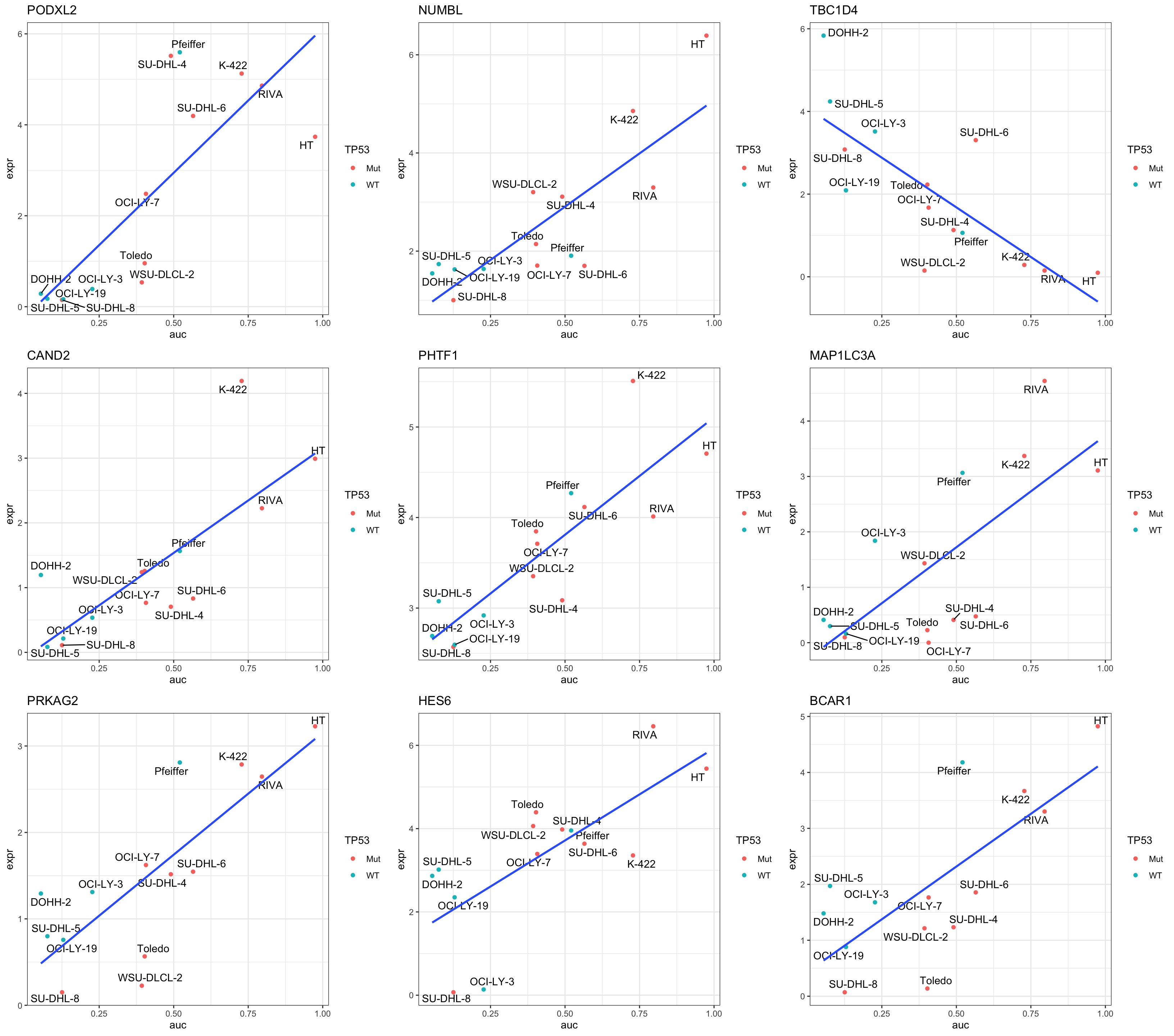
Enrichment analysis
gmts = list(H= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt",
C6 = "../data/gmts/c6.all.v6.2.symbols.gmt")
inputTab <- resTab %>% filter(P.Value < 0.1) %>%
distinct(symbol, .keep_all = TRUE) %>%
select(symbol, t) %>% data.frame() %>% column_to_rownames("symbol")
enRes <- list()
enRes[["Hallmark"]] <- runGSEA(inputTab, gmts$H, "page")
enRes[["KEGG"]] <- runGSEA(inputTab, gmts$KEGG,"page")
enRes[["Perturbation"]] <- runGSEA(inputTab, gmts$C6,"page")
p <- plotEnrichmentBar(enRes, pCut =0.01, ifFDR= FALSE)
cowplot::plot_grid(p)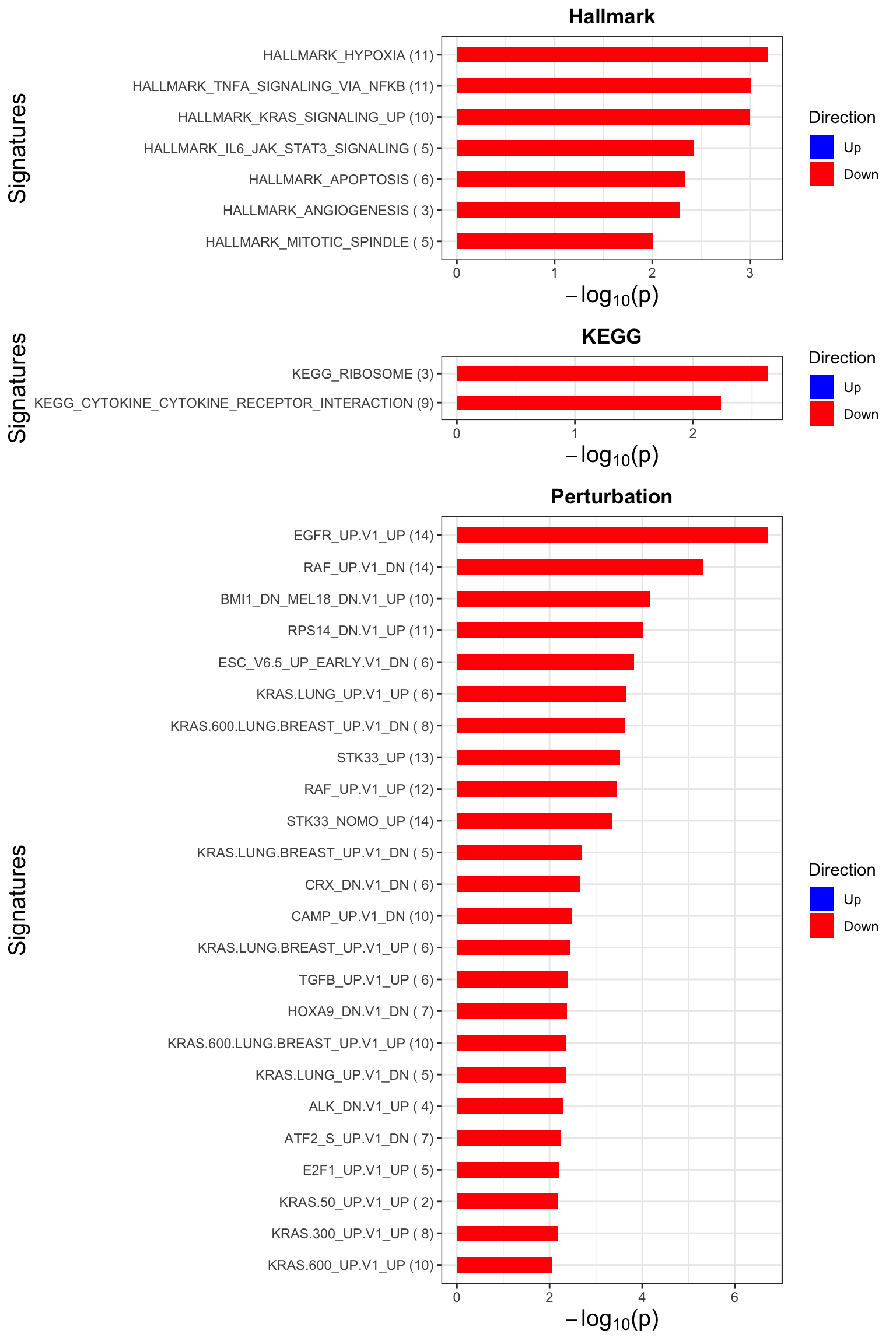
Focus on proteins from Fatty acid metabolism pathway
geneList <- piano::loadGSC(gmts$H)$gsc$HALLMARK_FATTY_ACID_METABOLISM
plotGene <- filter(filter(resTab, P.Value <= 0.05), symbol%in% geneList )
pList <- lapply(seq(nrow(plotGene)), function(i) {
rec <- plotGene[i,]
plotTab <- tibble(expr = exprMat[rec$symbol,],
auc = colTab$auc,
TP53 = colTab$TP53,
Name = colnames(exprMat))
ggplot(plotTab, aes(x=auc, y=expr)) +
geom_point(aes(col = TP53)) +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(rec$symbol) +
geom_smooth(method="lm", se= FALSE) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList,ncol=3)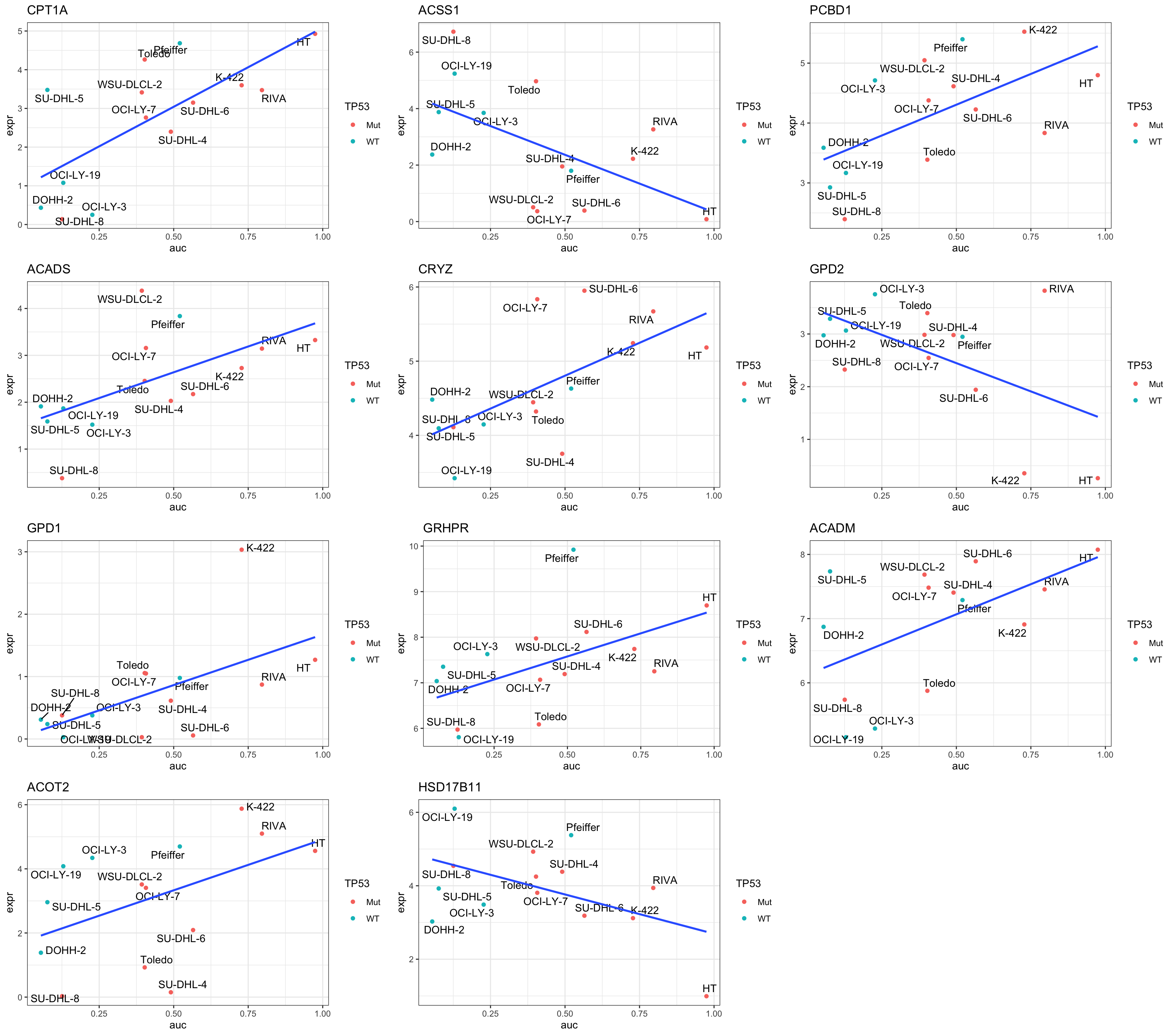
MOFA analysis
library(MOFA2)Preprocessing
mofaData <- omicListOnly use top 5000 most variant rna expression
exprMat <- mofaData$rna
sds <- genefilter::rowSds(exprMat)
exprMat <- exprMat[order(sds, decreasing = TRUE)[1:5000],]
mofaData$rna <- exprMatChange column names of the metabolite matrix
colnames(mofaData$metabolite) <- str_remove(colnames(mofaData$metabolite),"_U")Create MOFA object
# Create MultiAssayExperiment object
mofaData <- MultiAssayExperiment::MultiAssayExperiment(
experiments = mofaData
)MOFAobject <- create_mofa_from_MultiAssayExperiment(mofaData)Plot data overview
plot_data_overview(MOFAobject)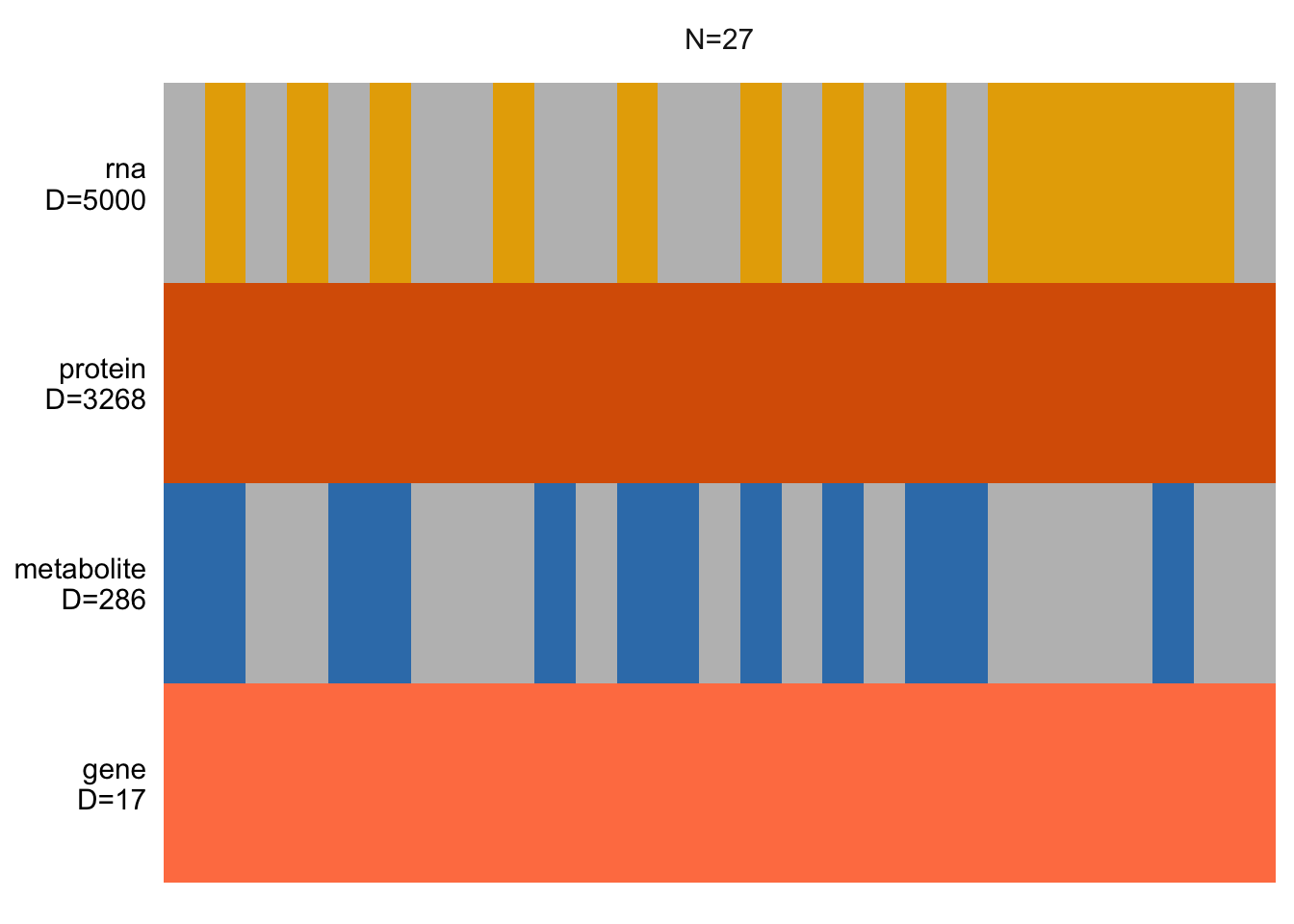
Define MOFA options
Data options
data_opts <- get_default_data_options(MOFAobject)
data_opts$scale_views
[1] FALSE
$scale_groups
[1] FALSE
$center_groups
[1] TRUE
$use_float32
[1] FALSE
$views
[1] "gene" "protein" "metabolite" "rna"
$groups
[1] "group1"Model options
model_opts <- get_default_model_options(MOFAobject)
model_opts$num_factors <- 10
model_opts$likelihoods
gene protein metabolite rna
"gaussian" "gaussian" "gaussian" "gaussian"
$num_factors
[1] 10
$spikeslab_factors
[1] FALSE
$spikeslab_weights
[1] TRUE
$ard_factors
[1] FALSE
$ard_weights
[1] TRUEChange the likely hood of Mutations to “bernoulli
model_opts$likelihoods[["gene"]] <- "bernoulli"
model_opts$likelihoods gene protein metabolite rna
"bernoulli" "gaussian" "gaussian" "gaussian" Training options
train_opts <- get_default_training_options(MOFAobject)
train_opts$convergence_mode <- "slow"
train_opts$seed <- 2022
train_opts$maxiter <- 10000
train_opts$maxiter
[1] 10000
$convergence_mode
[1] "slow"
$drop_factor_threshold
[1] -1
$verbose
[1] FALSE
$startELBO
[1] 1
$freqELBO
[1] 5
$stochastic
[1] FALSE
$gpu_mode
[1] FALSE
$seed
[1] 2022
$outfile
NULL
$weight_views
[1] FALSE
$save_interrupted
[1] FALSEChange drop threshold to 0.01
train_opts$drop_factor_threshold <-0.01Train the MOFA model
Prepare MOFA object
MOFAobject <- prepare_mofa(MOFAobject,
data_options = data_opts,
model_options = model_opts,
training_options = train_opts
)Training
MOFAobject <- run_mofa(MOFAobject)
saveRDS(MOFAobject,"../output/mofaDLBCL_Doxo.rds")Preliminary analysis of the results
MOFAobject <- readRDS("../output/mofaDLBCL_Doxo.rds")Factor correlation matrix
plot_factor_cor(MOFAobject)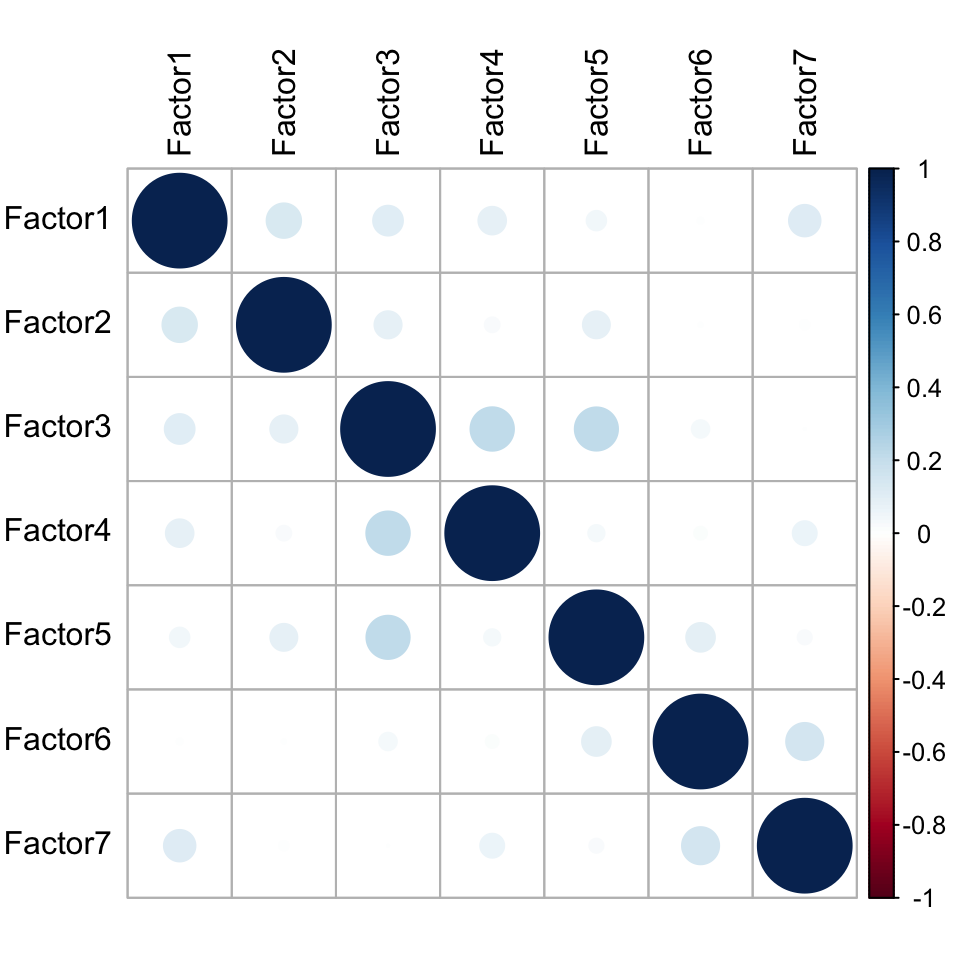
Variance explained
plot_variance_explained(MOFAobject, max_r2=15)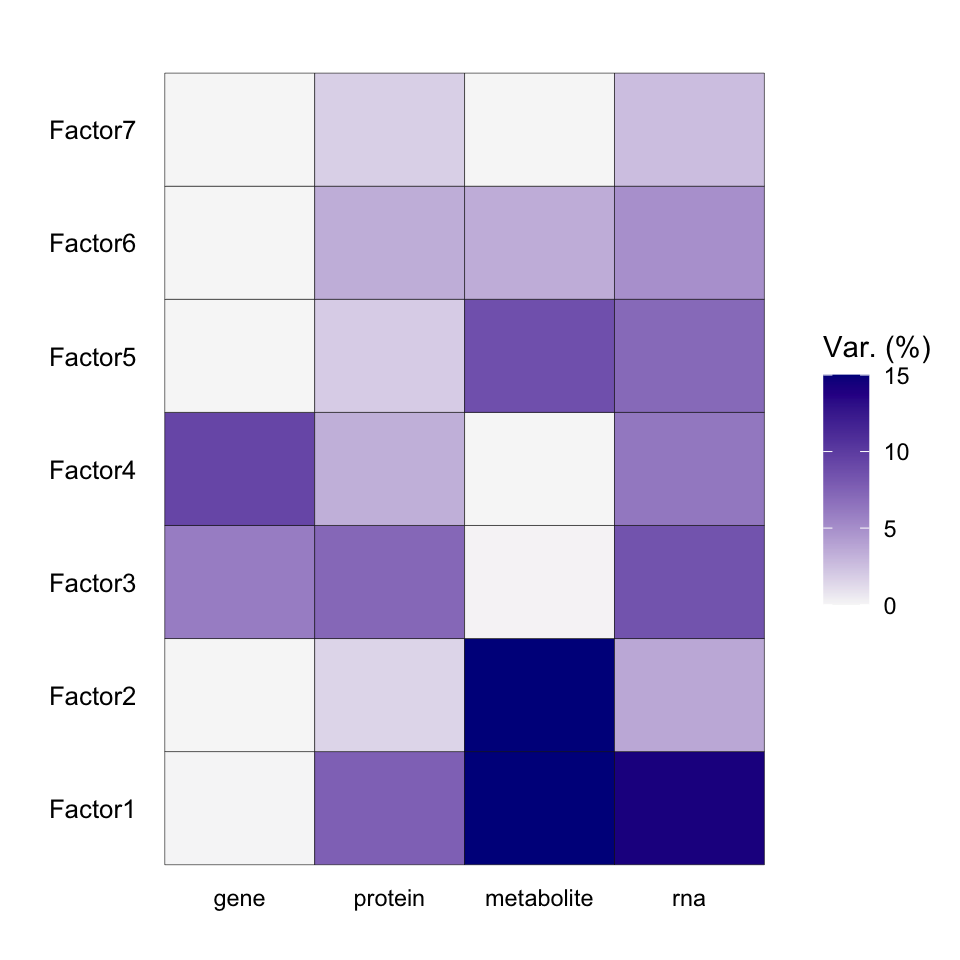
Total variance explained
plot_variance_explained(MOFAobject, plot_total = T)[[2]]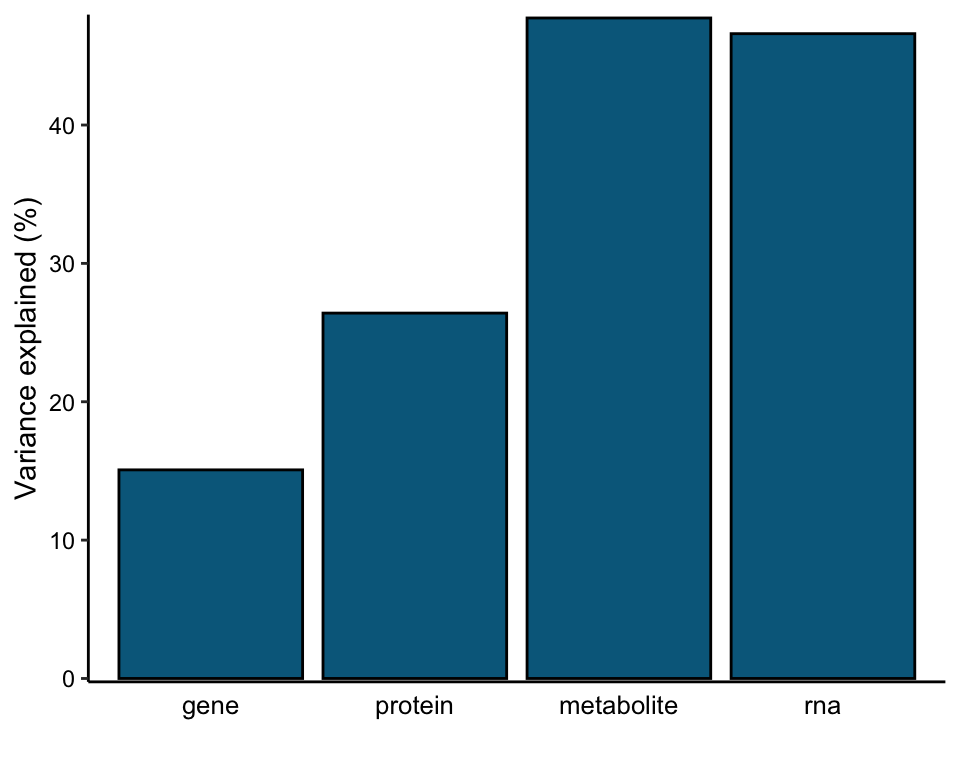
Factor heatmap
library(pheatmap)
#gene annotation
facMat <- t(get_factors(MOFAobject)[[1]])
colAnno <- aucTab %>% column_to_rownames("Name")
pheatmap(facMat, clustering_method = "ward.D2", annotation_col = colAnno)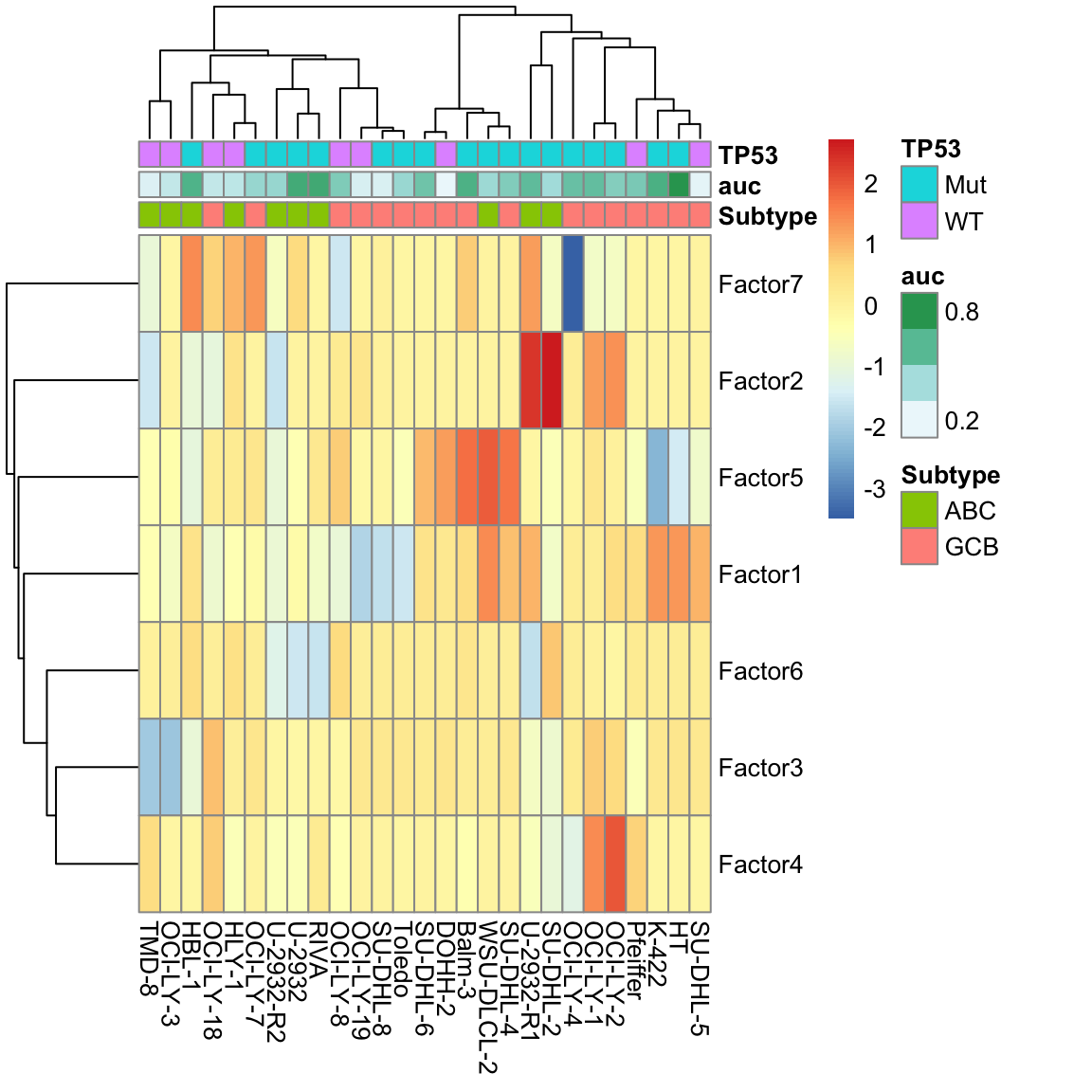
Correlation between factors and annotations
facTab <- facMat %>% as_tibble(rownames = "factor") %>%
pivot_longer(-factor, names_to = "Name", values_to = "value") %>%
left_join(aucTab, by= "Name")Subtype
resTab <- facTab %>% group_by(factor) %>% nest() %>%
mutate(m=map(data, ~t.test(value ~ Subtype,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(factor, estimate, p.value) %>%
arrange(p.value)
head(resTab)# A tibble: 6 × 3
# Groups: factor [6]
factor estimate p.value
<chr> <dbl> <dbl>
1 Factor3 -0.957 0.00518
2 Factor4 -0.380 0.100
3 Factor6 -0.555 0.112
4 Factor7 0.443 0.244
5 Factor5 -0.204 0.588
6 Factor1 -0.117 0.743 ggplot(filter(facTab, factor == "Factor3"), aes(x=Subtype, y=value)) +
geom_boxplot() + geom_point()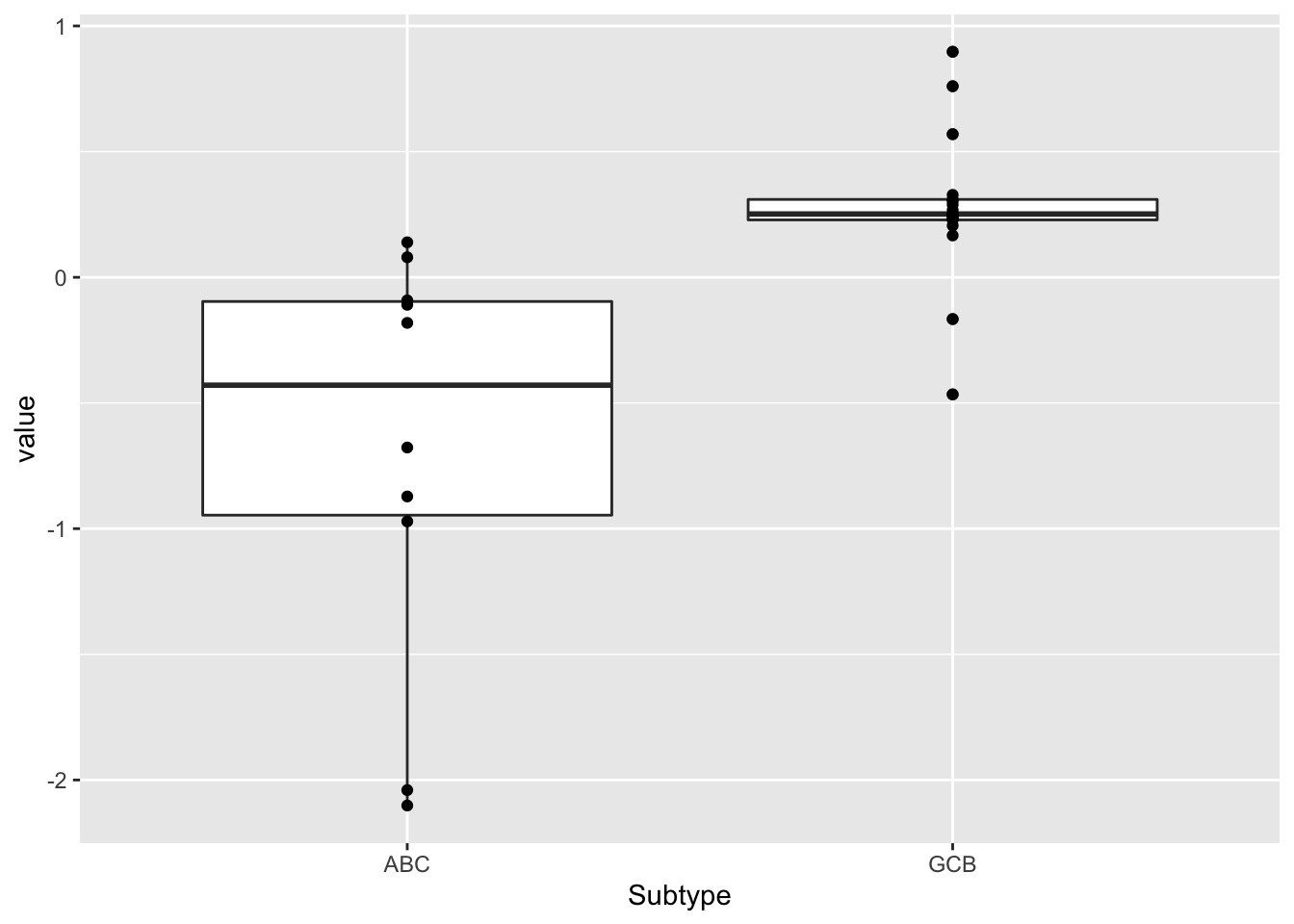
TP55
resTab <- facTab %>% group_by(factor) %>% nest() %>%
mutate(m=map(data, ~t.test(value ~ TP53,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(factor, estimate, p.value) %>%
arrange(p.value)
head(resTab)# A tibble: 6 × 3
# Groups: factor [6]
factor estimate p.value
<chr> <dbl> <dbl>
1 Factor6 -0.382 0.0594
2 Factor2 0.477 0.164
3 Factor1 0.484 0.201
4 Factor3 0.371 0.339
5 Factor4 -0.149 0.528
6 Factor7 0.0648 0.860 ggplot(filter(facTab, factor == "Factor6"), aes(x=TP53, y=value)) +
geom_boxplot() + geom_point()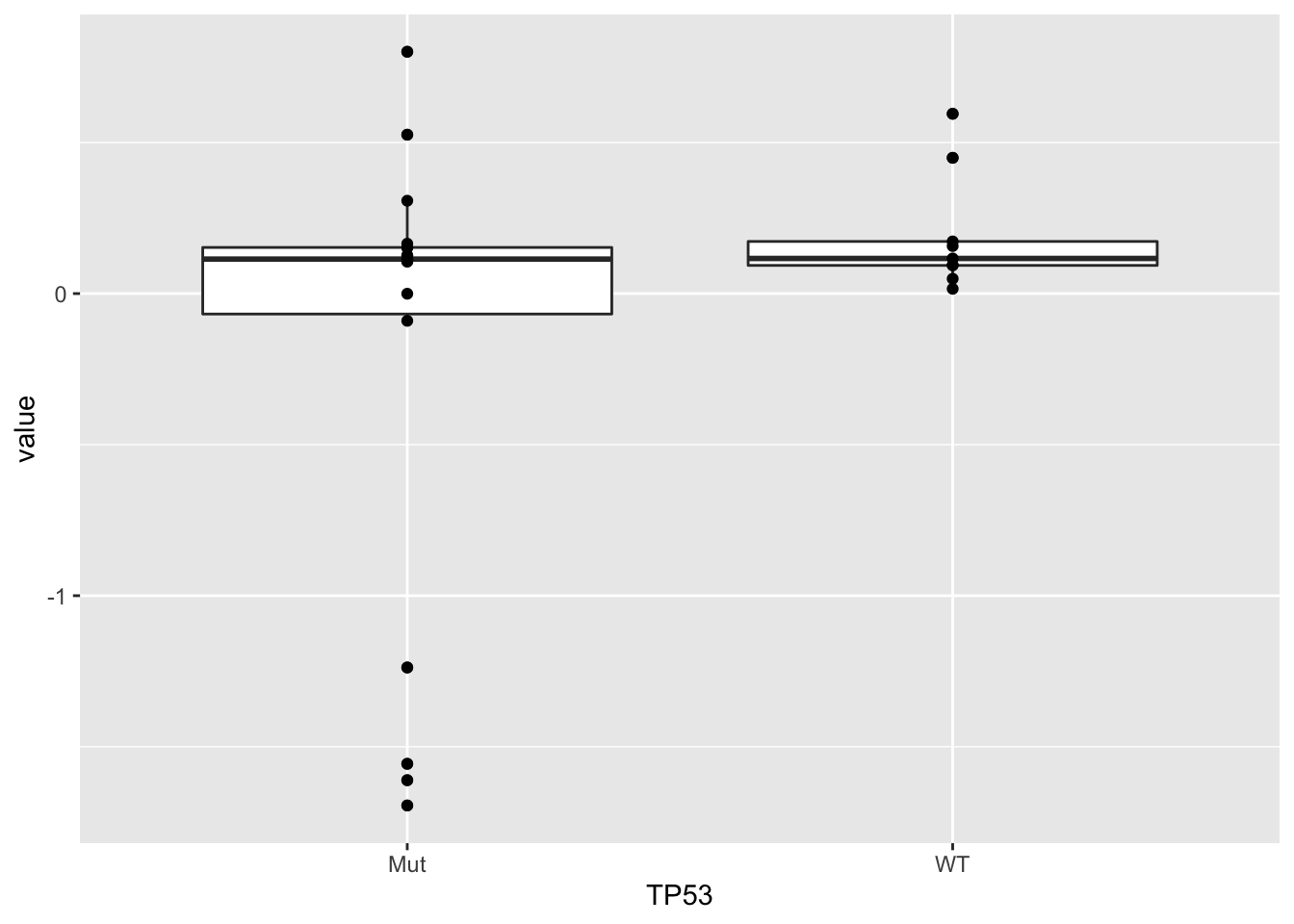
Doxorubicin response
resTab <- facTab %>% group_by(factor) %>% nest() %>%
mutate(m=map(data, ~cor.test(~ value + auc,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(factor, estimate, p.value) %>%
arrange(p.value)
head(resTab)# A tibble: 6 × 3
# Groups: factor [6]
factor estimate p.value
<chr> <dbl> <dbl>
1 Factor1 0.430 0.0251
2 Factor6 -0.311 0.114
3 Factor5 -0.170 0.397
4 Factor2 0.136 0.500
5 Factor3 0.127 0.529
6 Factor4 -0.0588 0.771 ggplot(filter(facTab, factor == "Factor1"), aes(x=auc, y=value)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
ggrepel::geom_text_repel(aes(label = Name))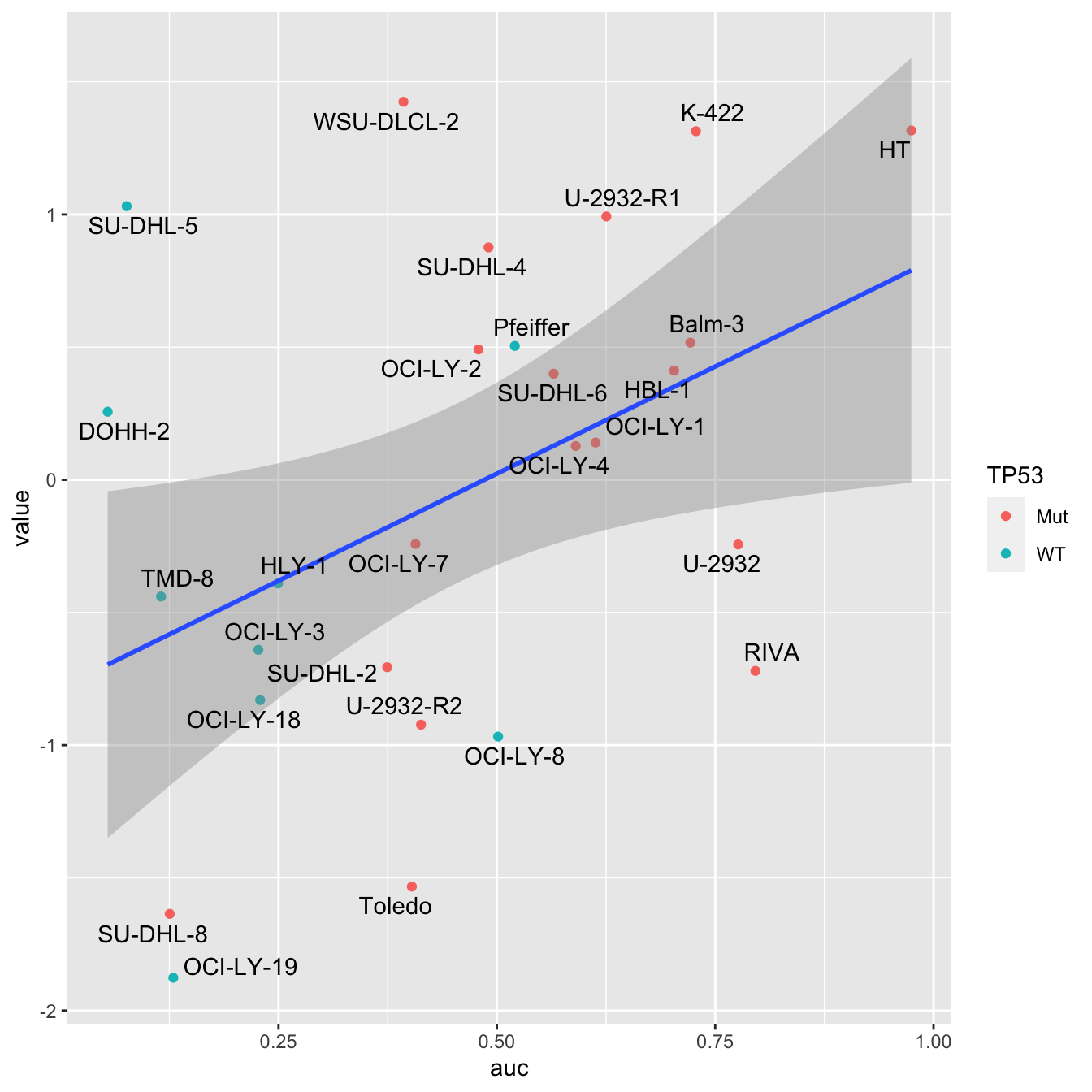
Focus on F1 (Doxorubicine response)
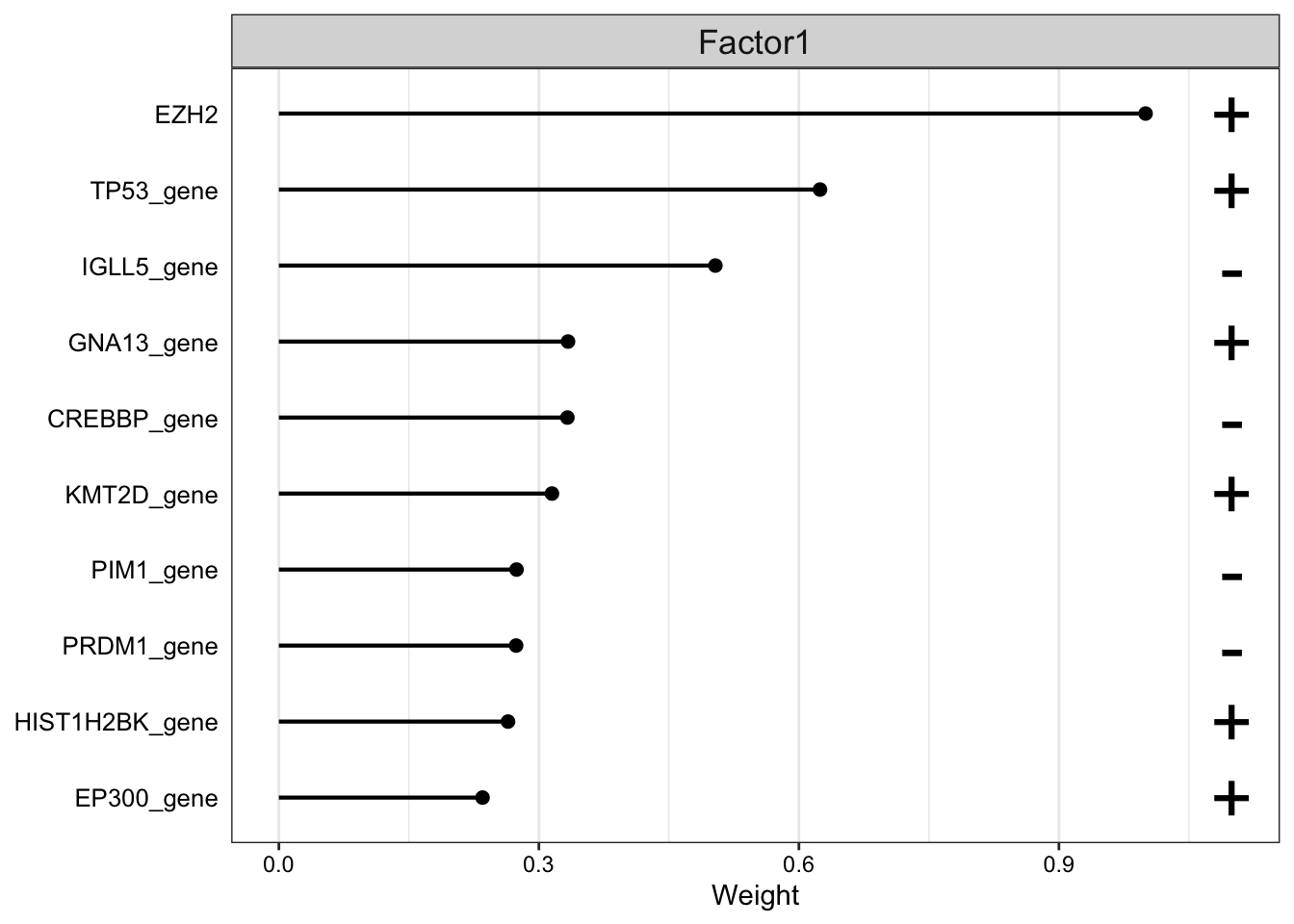
Weight of genomic features
plot_top_weights(MOFAobject,
view = "gene",
factor = 1,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
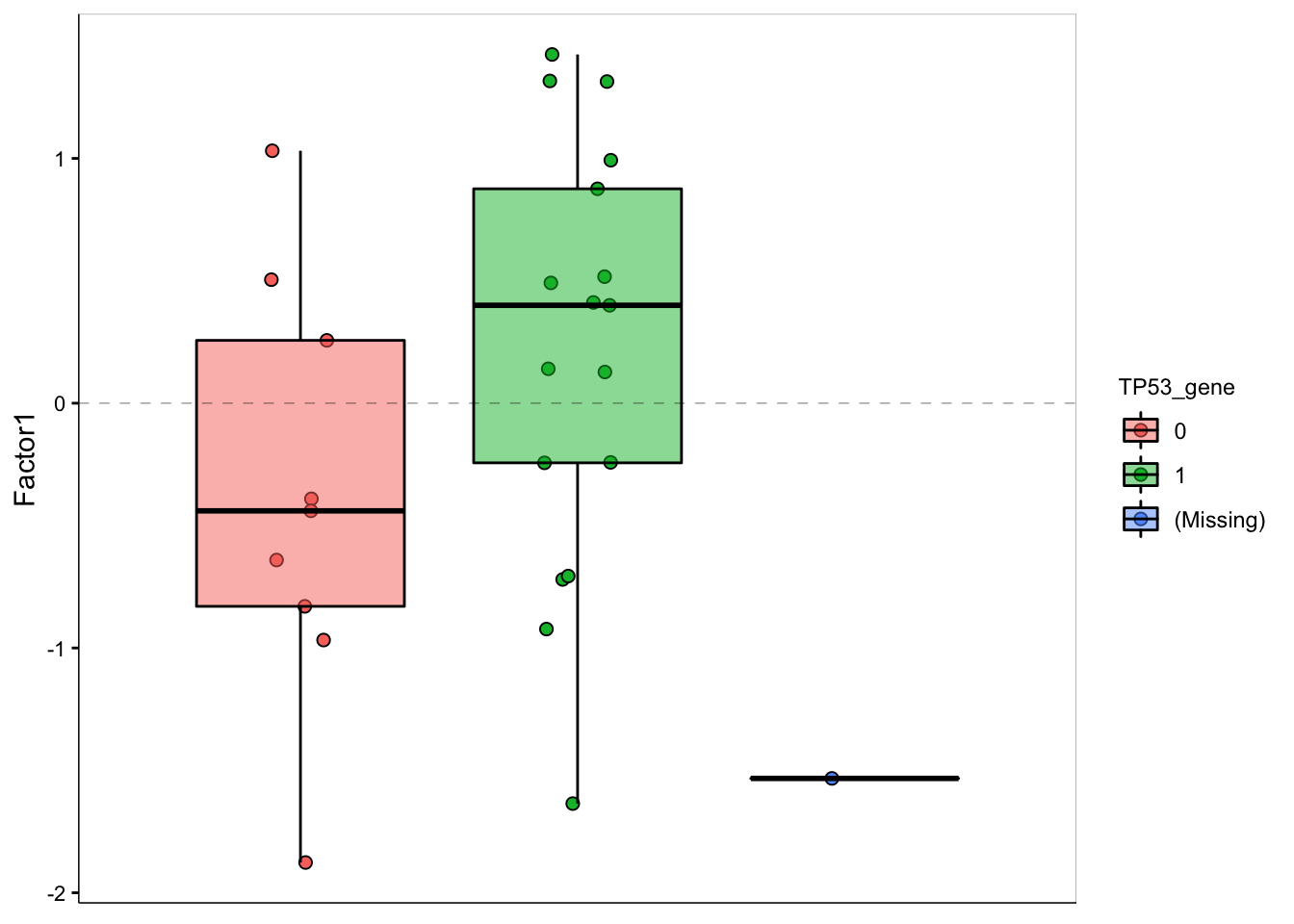
F1 values versus TP53 mutation
plot_factor(MOFAobject,
factors = 1,
color_by = "TP53_gene",
add_violin = TRUE,
dodge = TRUE
)
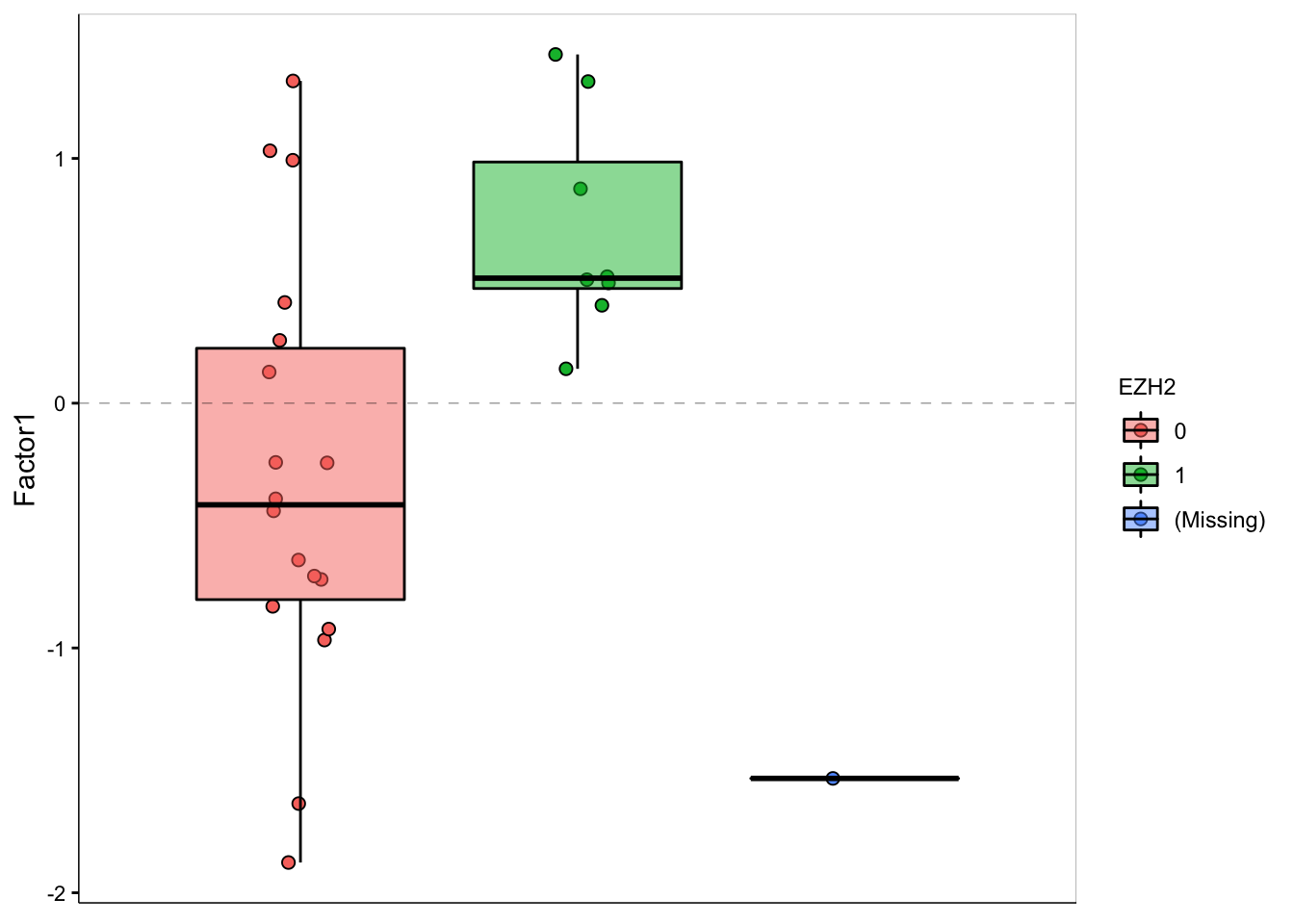
F1 values versus EZH2 mutation
plot_factor(MOFAobject,
factors = 1,
color_by = "EZH2",
add_violin = TRUE,
dodge = TRUE
)
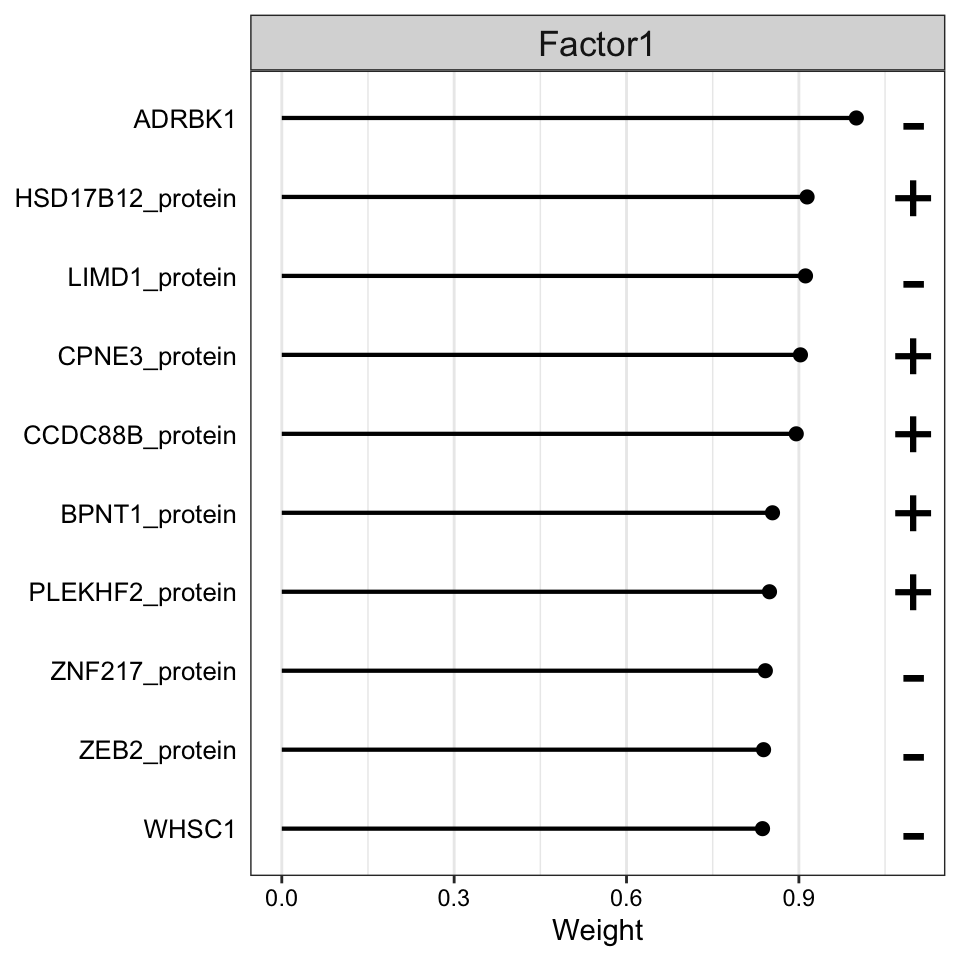
Weight of protein features
plot_top_weights(MOFAobject,
view = "protein",
factor = 1,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
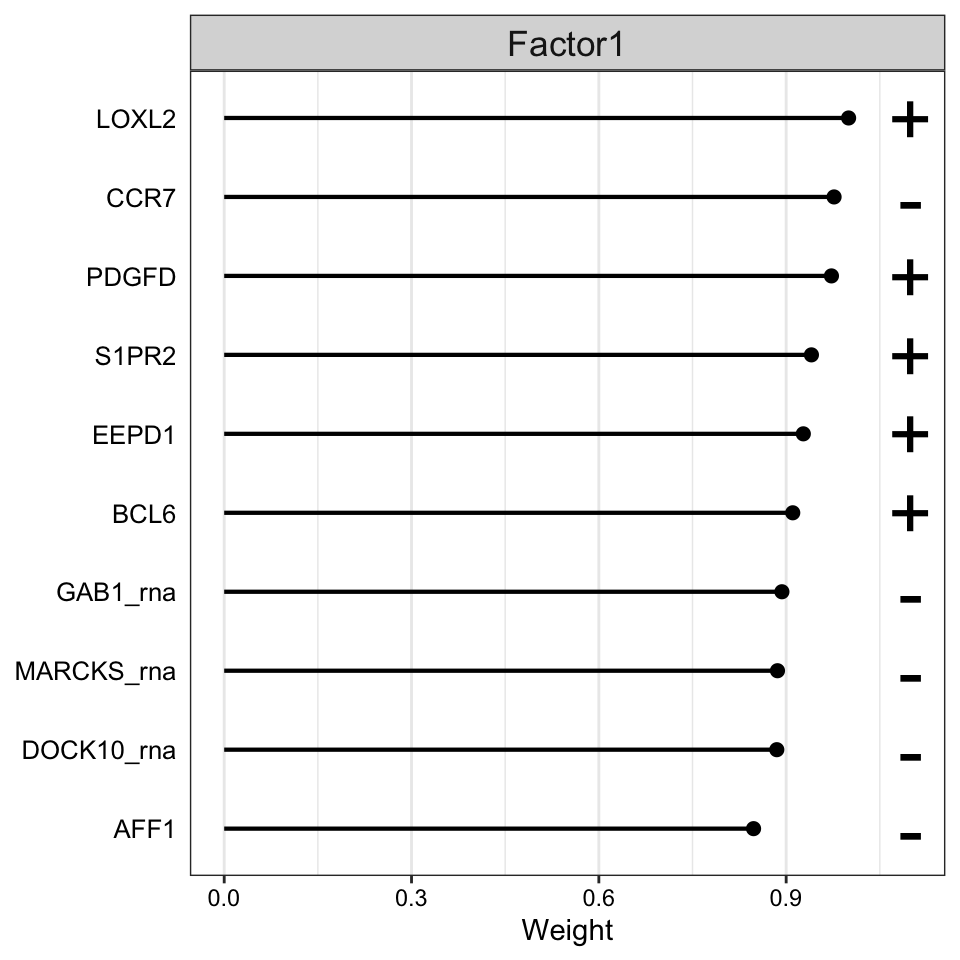
Weight of rna features
plot_top_weights(MOFAobject,
view = "rna",
factor = 1,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
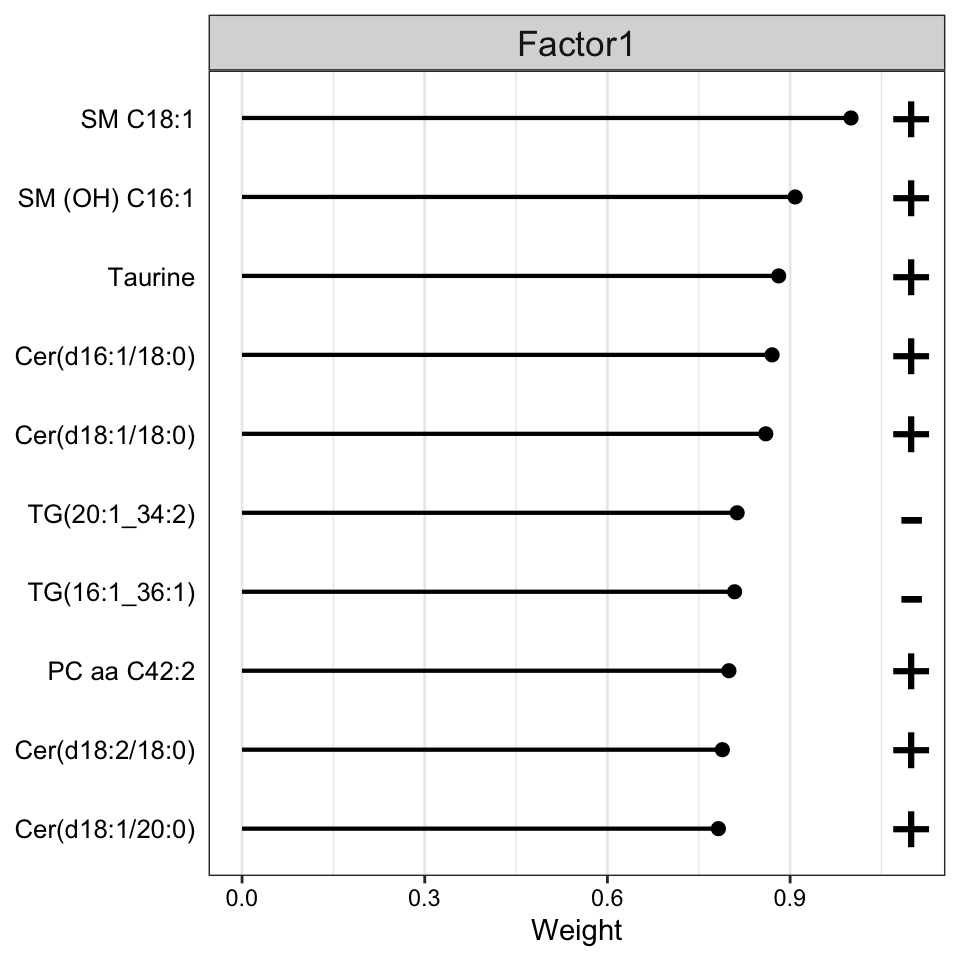
Weight of metabolites features
plot_top_weights(MOFAobject,
view = "metabolite",
factor = 1,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
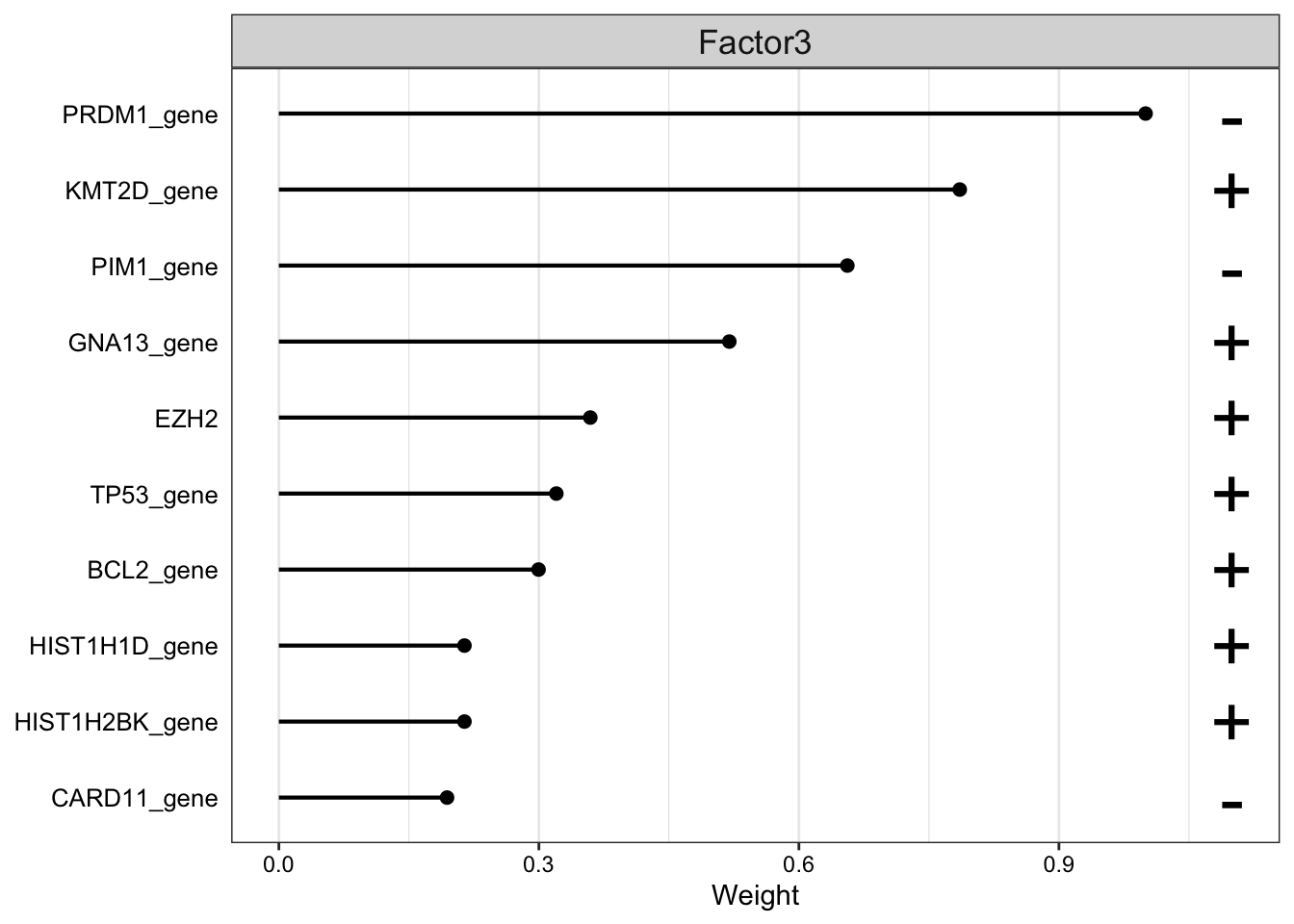
Focus on F3 (Subtype)
Weight of genomic features on LF3
plot_top_weights(MOFAobject,
view = "gene",
factor =3,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
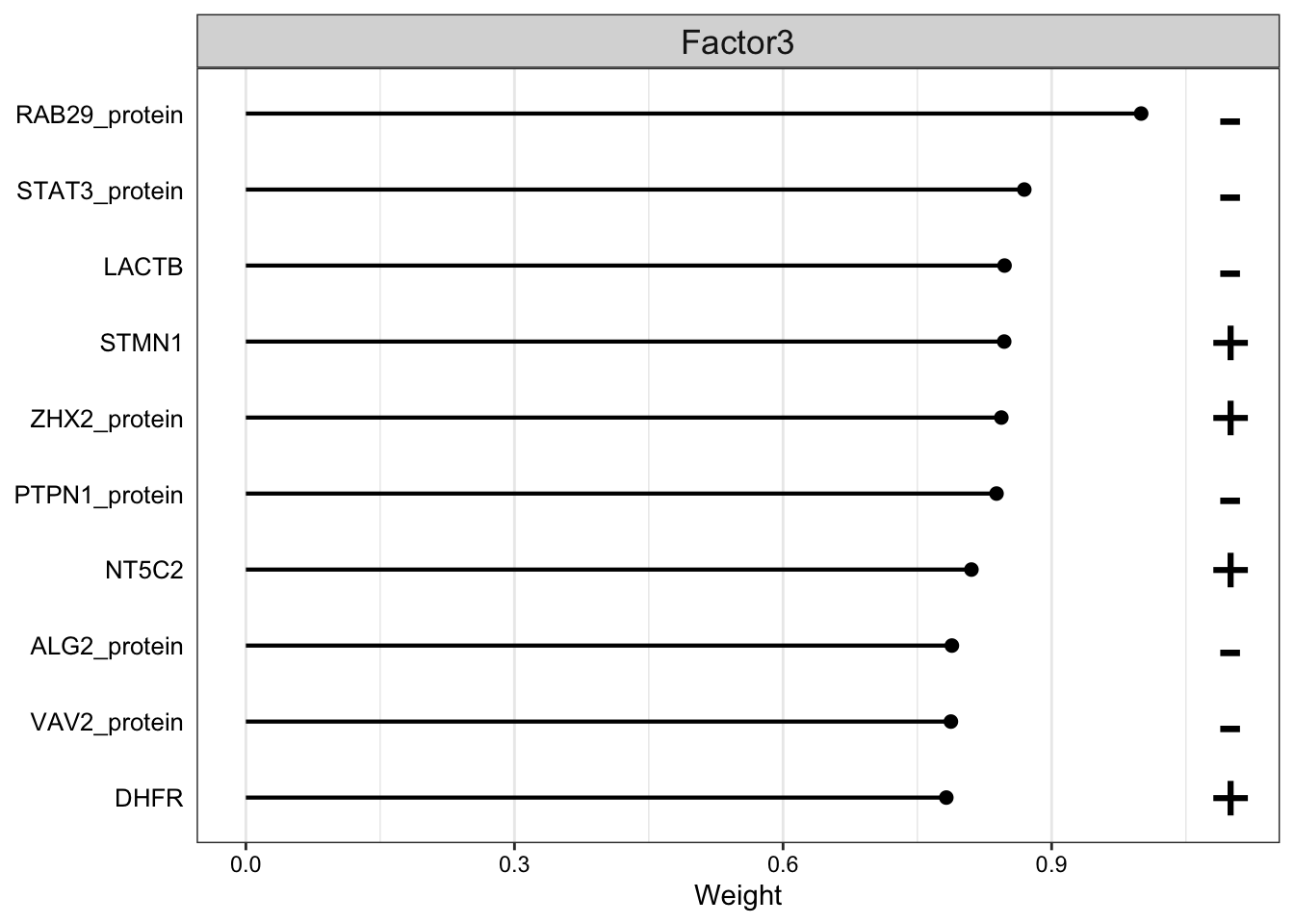
Weight of protein features on LF3
plot_top_weights(MOFAobject,
view = "protein",
factor =3,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
Weight of rna features on LF3
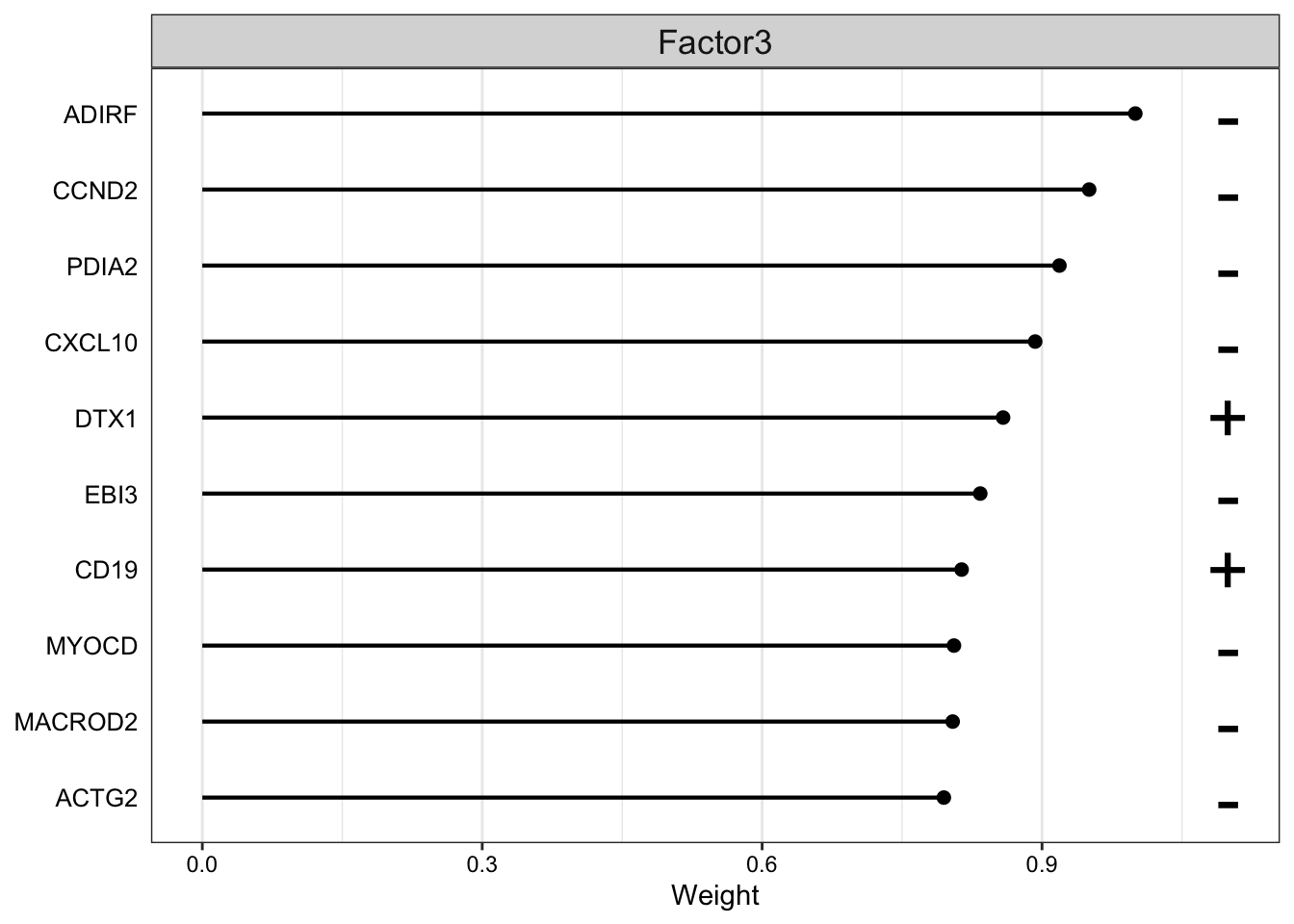
plot_top_weights(MOFAobject,
view = "rna",
factor =3,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
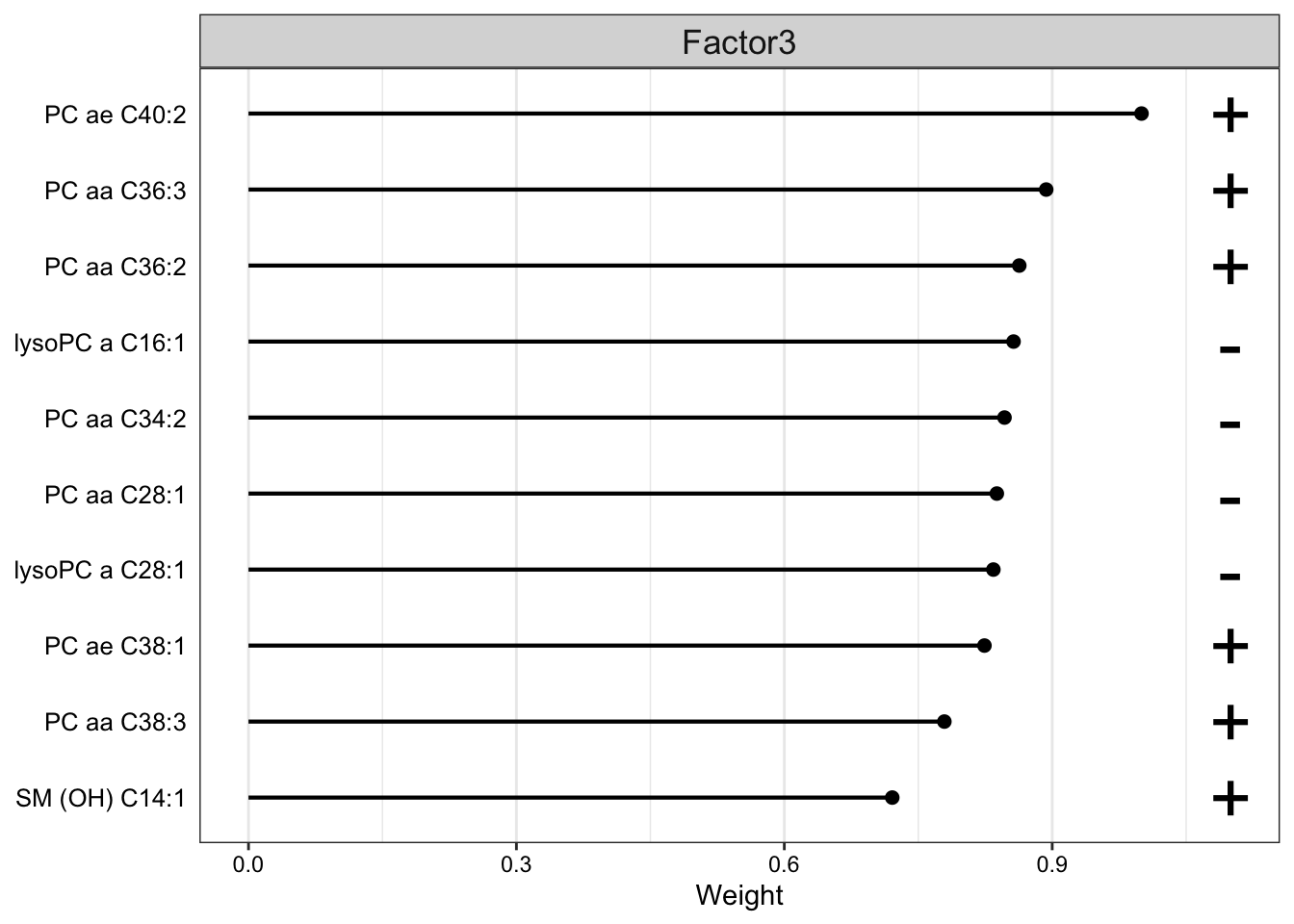
Weight of metabolite features on LF3
plot_top_weights(MOFAobject,
view = "metabolite",
factor =3,
nfeatures = 10, # Top number of features to highlight
scale = T # Scale weights from -1 to 1
)
LASSO regression for predicting Doxorubicine responses
library(glmnet)
detach(package:MOFA2, unload = TRUE)Preprocessing data
Response vector
responseList <- list(Doxorubicin = structure(aucTab$auc, names=aucTab$Name))RNAseq data
lassoData <- omicList
exprMat <- lassoData$rna
sds <- genefilter::rowSds(exprMat)
exprMat <- exprMat[order(sds, decreasing = TRUE)[1:5000],]
exprMat <- t(exprMat)
colnames(exprMat) <- paste0(colnames(exprMat),"_rna")Proteomic data
protMat <- t(lassoData$protein)
colnames(protMat) <- paste0(colnames(protMat),"_protein")Genomic data
geneMat <- t(lassoData$gene)
colnames(geneMat) <- paste0(colnames(geneMat),"_gene")Metabolomic data
metaMat <- lassoData$metabolite
colnames(metaMat) <- str_remove(colnames(metaMat),"_U")
metaMat <- t(metaMat)
colnames(metaMat) <- paste0(colnames(metaMat),"_metabolite")Feature selection with LASSO penalty
#Functions for running glm
runGlm <- function(X, y, method = "ridge", repeats=20, folds = 3, lambda = "lambda.min") {
modelList <- list()
lambdaList <- c()
varExplain <- c()
coefMat <- matrix(NA, ncol(X), repeats)
rownames(coefMat) <- colnames(X)
if (method == "lasso"){
alpha = 1
} else if (method == "ridge") {
alpha = 0
}
for (i in seq(repeats)) {
if (ncol(X) > 2) {
res <- cv.glmnet(X,y, type.measure = "mse", family="gaussian",
nfolds = folds, alpha = alpha, standardize = FALSE)
lambdaList <- c(lambdaList, res[[lambda]])
modelList[[i]] <- res
coefModel <- coef(res, s = lambda)[-1] #remove intercept row
coefMat[,i] <- coefModel
#calculate variance explained
y.pred <- predict(res, s = lambda, newx = X)
varExp <- cor(as.vector(y),as.vector(y.pred))^2
varExplain[i] <- ifelse(is.na(varExp), 0, varExp)
} else {
fitlm<-lm(y~., data.frame(X))
varExp <- summary(fitlm)$r.squared
varExplain <- c(varExplain, varExp)
}
}
list(modelList = modelList, lambdaList = lambdaList, varExplain = varExplain, coefMat = coefMat)
}#function for scaling predictors
dataScale <- function(x, censor = NULL, robust = FALSE) {
#function to scale different variables
if (length(unique(na.omit(x))) <=3){
#a binary variable, change to -0.5 and 0.5 for 1 and 2
x - 0.5
} else {
if (robust) {
#continuous variable, centered by median and divied by 2*mad
mScore <- (x-median(x,na.rm=TRUE))/mad(x,na.rm=TRUE)
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
} else {
mScore <- (x-mean(x,na.rm=TRUE))/(sd(x,na.rm=TRUE))
if (!is.null(censor)) {
mScore[mScore > censor] <- censor
mScore[mScore < -censor] <- -censor
}
mScore/2
}
}
}#function to generate response vector and explainatory variable for each seahorse measurement
generateData <- function(responseList, inclSet, onlyCombine = FALSE, censor = NULL, robust = FALSE) {
allResponse <- list()
allExplain <- list()
for (measure in names(responseList)) {
y <- responseList[[measure]]
y <- y[!is.na(y)]
#get overlapped samples for each dataset
overSample <- names(y)
for (eachSet in inclSet) {
overSample <- intersect(overSample,rownames(eachSet))
}
y <- dataScale(y[overSample], censor = censor, robust = robust)
expTab <- list()
if ("Gene" %in% names(inclSet)) {
geneTab <- inclSet$Gene[overSample,]
#at least 3 mutated sample
geneTab <- geneTab[, colSums(geneTab) >= 3]
vecName <- sprintf("genetic(%s)", ncol(geneTab))
expTab[[vecName]] <- apply(geneTab,2,dataScale)
}
if ("RNA" %in% names(inclSet)){
rnaMat <- inclSet$RNA[overSample, ]
colnames(rnaMat) <- paste0("con.",colnames(rnaMat), sep = "")
vecName <- sprintf("RNA(%s)", ncol(rnaMat))
expTab[[vecName]] <- apply(rnaMat,2,dataScale, censor = censor, robust = robust)
}
if ("Protein" %in% names(inclSet)){
protMat <- inclSet$Protein[overSample, ]
colnames(protMat) <- paste0("con.",colnames(protMat), sep = "")
vecName <- sprintf("Protein(%s)", ncol(protMat))
expTab[[vecName]] <- apply(protMat,2,dataScale, censor = censor, robust = robust)
}
if ("Metabolite" %in% names(inclSet)){
metaMat <- inclSet$Metabolite[overSample, ]
colnames(metaMat) <- paste0("con.",colnames(metaMat), sep = "")
vecName <- sprintf("Metabolite(%s)", ncol(metaMat))
expTab[[vecName]] <- apply(metaMat,2,dataScale, censor = censor, robust = robust)
}
if (length(inclSet) > 1) {
comboTab <- c()
for (eachSet in names(expTab)){
comboTab <- cbind(comboTab, expTab[[eachSet]])
}
vecName <- sprintf("all(%s)", ncol(comboTab))
expTab[[vecName]] <- comboTab
}
allResponse[[measure]] <- y
allExplain[[measure]] <- expTab
}
if (onlyCombine) {
#only return combined results, for feature selection
allExplain <- lapply(allExplain, function(x) x[length(x)])
}
return(list(allResponse=allResponse, allExplain=allExplain))
}
italicizeGene <- function(featureNames) {
geneNameList <- c("SF3B1","NOTCH1","TP53", "DDX3X","MED12", "ATM","BRAF","EGR2")
specialCase <- "TP53/del17p"
formatedList <- sapply(featureNames, function(n) {
if (n %in% geneNameList) {
sprintf("italic('%s')",n)
} else if (n == specialCase) {
sprintf("italic('TP53')~'/del17p'")
}
else {
sprintf("'%s'",n)
}
})
return(formatedList)
}
formatCNV <- function(cnvList) {
nameCNV <- c(del17p = "del(17)(p13)", del11q = "del(11)(q22.3)", del13q = "del(13)(q14)", trisomy12 = "trisomy 12", trisomy19 = "trisomy 19")
cnvList <- sapply(cnvList, function(n) {
if (n %in% names(nameCNV)) {
n <- nameCNV[n]
}
n
})
cnvList
}
library(gtable)
lassoPlot <- function(lassoOut, cleanData, freqCut = 1, coefCut = 0.01,
setNumber = "last", legend = TRUE, labSuffix = " response", scaleFac =1) {
plotList <- list()
if (setNumber == "last") {
setNumber <- length(lassoOut[[1]])
} else {
setNumber <- setNumber
}
for (seaName in names(lassoOut)) {
#for the barplot on the left of the heatmap
barValue <- rowMeans(lassoOut[[seaName]][[setNumber]]$coefMat)
freqValue <- rowMeans(abs(sign(lassoOut[[seaName]][[setNumber]]$coefMat)))
barValue <- barValue[abs(barValue) >= coefCut & freqValue >= freqCut] # a certain threshold
barValue <- barValue[order(barValue)]
if(length(barValue) == 0) {
plotList[[seaName]] <- NA
next
}
#for the heatmap and scatter plot below the heatmap
allData <- cleanData$allExplain[[seaName]][[setNumber]]
seaValue <- cleanData$allResponse[[seaName]]*2 #back to Z-score
tabValue <- allData[, names(barValue),drop=FALSE]
ord <- order(seaValue)
seaValue <- seaValue[ord]
tabValue <- tabValue[ord, ,drop=FALSE]
sampleIDs <- rownames(tabValue)
tabValue <- as.tibble(tabValue)
#change scaled binary back to catagorical
for (eachCol in colnames(tabValue)) {
if (strsplit(eachCol, split = "[.]")[[1]][1] != "con") {
tabValue[[eachCol]] <- as.integer(as.factor(tabValue[[eachCol]]))
}
else {
tabValue[[eachCol]] <- tabValue[[eachCol]]*2 #back to Z-score
}
}
tabValue$Sample <- sampleIDs
#Mark different rows for different scaling in heatmap
matValue <- gather(tabValue, key = "Var",value = "Value", -Sample)
matValue$Type <- "mut"
#For continuious value
matValue$Type[grep("con.",matValue$Var)] <- "con"
#for methylation_cluster
matValue$Type[grep("ConsCluster",matValue$Var)] <- "meth"
#change the scale of the value, let them do not overlap with each other
matValue[matValue$Type == "mut",]$Value = matValue[matValue$Type == "mut",]$Value + 10
matValue[matValue$Type == "meth",]$Value = matValue[matValue$Type == "meth",]$Value + 20
#color scale for viability
idx <- matValue$Type == "con"
myCol <- colorRampPalette(c("blue",'white',"red"),
space = "Lab")
if (sum(idx) != 0) {
matValue[idx,]$Value = round(matValue[idx,]$Value,digits = 2)
minViab <- min(matValue[idx,]$Value)
maxViab <- max(matValue[idx,]$Value)
limViab <- max(c(abs(minViab), abs(maxViab)))
scaleSeq1 <- round(seq(-limViab, limViab,0.01), digits=2)
color4viab <- setNames(myCol(length(scaleSeq1+1)), nm=scaleSeq1)
} else {
scaleSeq1 <- round(seq(0,1,0.01), digits=2)
color4viab <- setNames(myCol(length(scaleSeq1+1)), nm=scaleSeq1)
}
#change continues measurement to discrete measurement
matValue$Value <- factor(matValue$Value,levels = sort(unique(matValue$Value)))
#change order of heatmap
names(barValue) <- gsub("con.", "", names(barValue))
matValue$Var <- gsub("con.","",matValue$Var)
matValue$Var <- factor(matValue$Var, levels = names(barValue))
matValue$Sample <- factor(matValue$Sample, levels = names(seaValue))
matValue <- matValue %>% mutate(varForm = as.character(Var)) %>%
mutate(varForm = formatCNV(varForm)) %>%
mutate(varForm = italicizeGene(varForm)) %>%
arrange(Var) %>% mutate(varForm = factor(varForm, levels = unique(varForm)))
#plot the heatmap
p1 <- ggplot(matValue, aes(x=Sample, y=Var)) + geom_tile(aes(fill=Value), color = "gray") +
theme_bw() +
scale_y_discrete(expand=c(0,0),position = "right",labels = parse(text = levels(matValue$varForm))) +
theme(axis.text.y=element_text(hjust=0, size=10*scaleFac), axis.text.x=element_blank(),
axis.title = element_blank(),
axis.ticks=element_blank(), panel.border=element_rect(colour="gainsboro"),
plot.title=element_blank(), panel.background=element_blank(),
panel.grid.major=element_blank(), panel.grid.minor=element_blank(),
plot.margin = margin(0,0,0,0)) +
scale_fill_manual(name="Mutated", values=c(color4viab, `11`="gray96", `12`='black', `21`='lightgreen',
`22`='green',`23` = 'green4'),guide=FALSE) #+ ggtitle(seaName)
#Plot the bar plot on the left of the heatmap
barDF = data.frame(barValue, nm=factor(names(barValue),levels=names(barValue)))
p2 <- ggplot(data=barDF, aes(x=nm, y=barValue)) +
geom_bar(stat="identity", fill="lightblue", colour="black", position = "identity", width=.66, size=0.2) +
theme_bw() + geom_hline(yintercept=0, size=0.3) + scale_x_discrete(expand=c(0,0.5)) +
scale_y_continuous(expand=c(0,0)) + coord_flip() +
theme(panel.grid.major=element_blank(), panel.background=element_blank(), axis.ticks.y = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x =element_text(size=8*scaleFac),
axis.text.y = element_blank(),
axis.title = element_blank(),
panel.border=element_blank(),plot.margin = margin(0,0,0,0)) + geom_vline(xintercept=c(0.5), color="black", size=0.6)
#Plot the scatter plot under the heatmap
# scatterplot below
scatterDF = data.frame(X=factor(names(seaValue), levels=names(seaValue)), Y=seaValue)
p3 <- ggplot(scatterDF, aes(x=X, y=Y)) + geom_point(shape=21, fill="dimgrey", colour="black", size=1.2) +
xlab(paste0(seaName, labSuffix)) + ylab("Z-score") +
theme_bw() +
theme(panel.grid.minor=element_blank(), panel.grid.major.x=element_blank(),
axis.title=element_text(size=10*scaleFac),
axis.text.x=element_blank(), axis.ticks.x=element_blank(),
axis.text.y=element_text(size=8*scaleFac),
panel.border=element_rect(colour="dimgrey", size=0.1),
panel.background=element_rect(fill="gray96"),plot.margin = margin(0,0,0,0))
dummyGrob <- ggplot() + theme_void()
#Scale bar for continuous variable
if (legend) {
Vgg = ggplot(data=data.frame(x=1, y=as.numeric(names(color4viab))), aes(x=x, y=y, color=y)) + geom_point() +
scale_color_gradientn(name="Z-score", colours =color4viab) +
theme(legend.title=element_text(size=12*scaleFac), legend.text=element_text(size=10*scaleFac))
barLegend <- plot_grid(gtable_filter(ggplotGrob(Vgg), "guide-box"))
#Assemble all the plots togehter
} else {
barLegend <- dummyGrob
}
gt <- egg::ggarrange(p2,p1,barLegend,dummyGrob, p3, dummyGrob, ncol=3, nrow=2,
widths = c(0.6,2,0.3), padding = unit(0,"line"), clip = "off",
heights = c(length(unique(matValue$Var))/2,2),draw = FALSE)
plotList[[seaName]] <- gt
}
return(plotList)
}
plotVar <- function(glmResult) {
pList <- lapply(names(glmResult), function(n) {
plotTab <- lapply(names(glmResult[[n]]), function(x) {
tibble(variable = x, value = glmResult[[n]][[x]]$varExplain)}) %>%
bind_rows() %>% group_by(variable) %>%
summarise(mean=mean(value, na.rm = TRUE),sd=sd(value, na.rm=TRUE))
ggplot(plotTab,aes(x=variable, y=mean, fill= variable)) +
geom_bar(position=position_dodge(), stat="identity", width = 0.8, col="black") +
geom_errorbar(aes(ymin=mean-sd, ymax=mean+sd, width = 0.3), position=position_dodge(.9)) +
theme_classic() + theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5, size =12),
plot.title = element_text(hjust =0.5),
axis.text.y = element_text(size=12),
axis.title = element_text(size=15),
legend.position = "none") +
scale_fill_brewer("Set1",type = "qual") + coord_cartesian(ylim = c(0,1)) +
ylab("R2") + xlab("") + ggtitle("")
})
pList
}Only genetics as predictors
Clean and integrate multi-omics data
inclSet<-list(Gene=geneMat)
cleanData <- generateData(responseList, inclSet, censor = 5)Run lasso
set.seed(2021)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
Variance explained
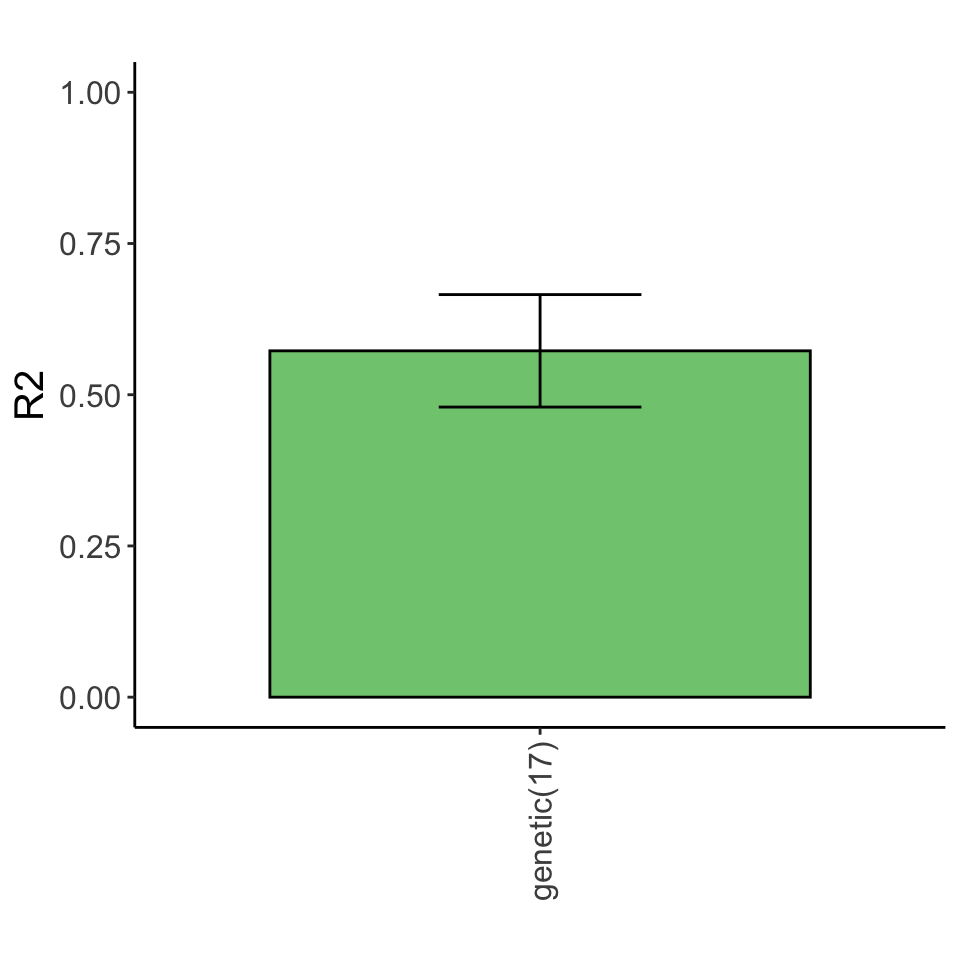
Lasso plot
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 0.8,setNumber = 1, legend = FALSE, scaleFac = 1)
heatMaps$Doxorubicin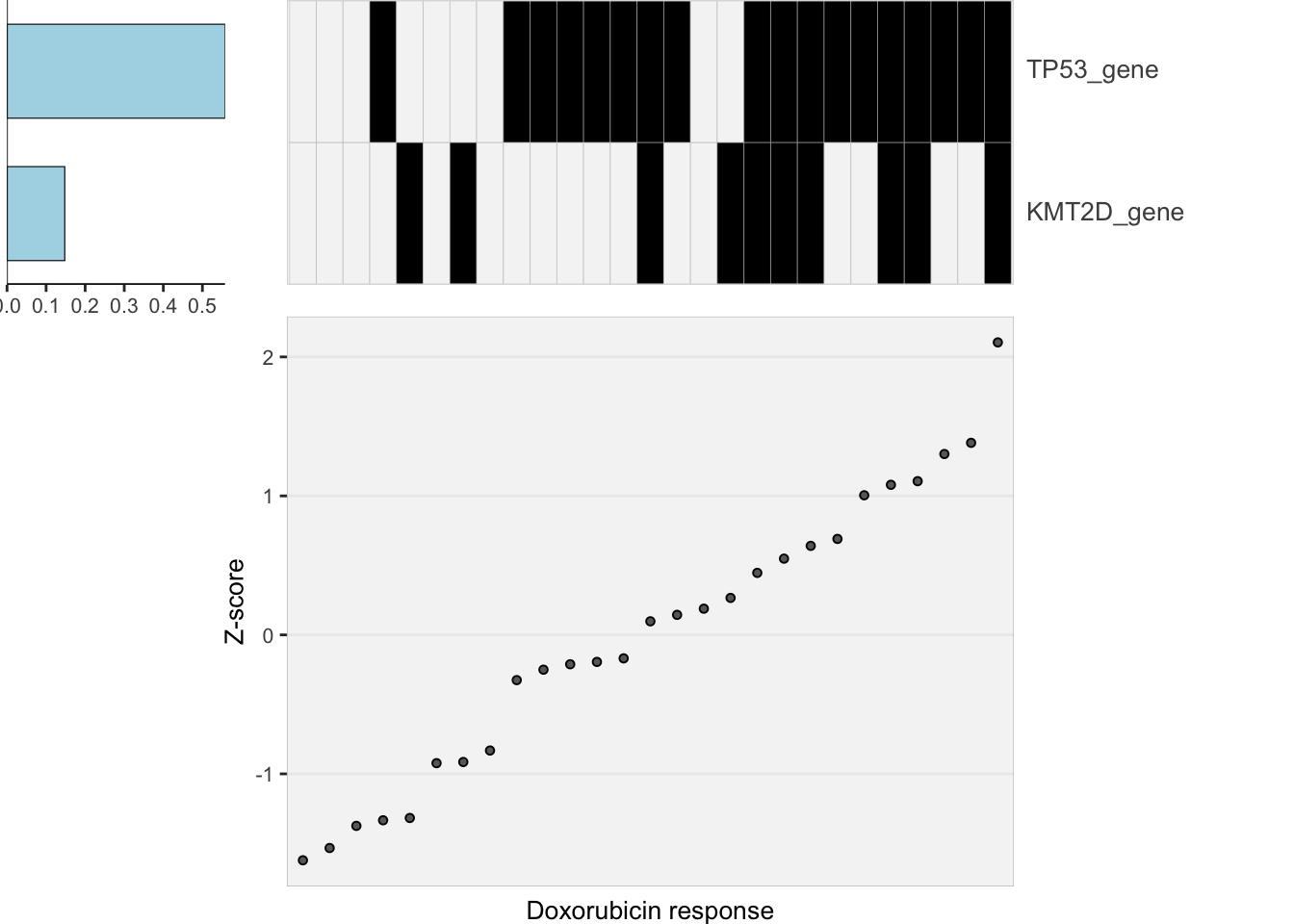
Only protein as predictors
Clean and integrate multi-omics data
inclSet<-list(Protein=protMat)
cleanData <- generateData(responseList, inclSet, censor = 5)Run lasso
set.seed(2021)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
Variance explained
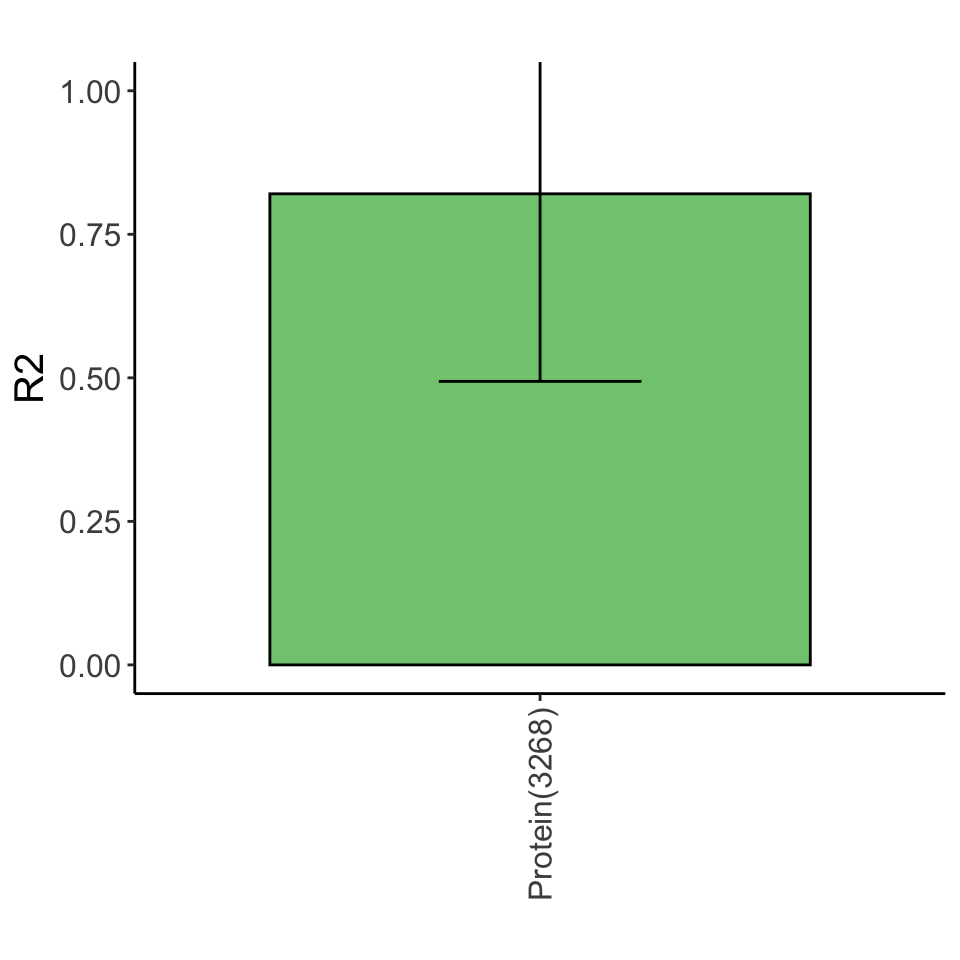
Lasso plot
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 0.8,setNumber = 1, legend = FALSE, scaleFac = 1)
heatMaps$Doxorubicin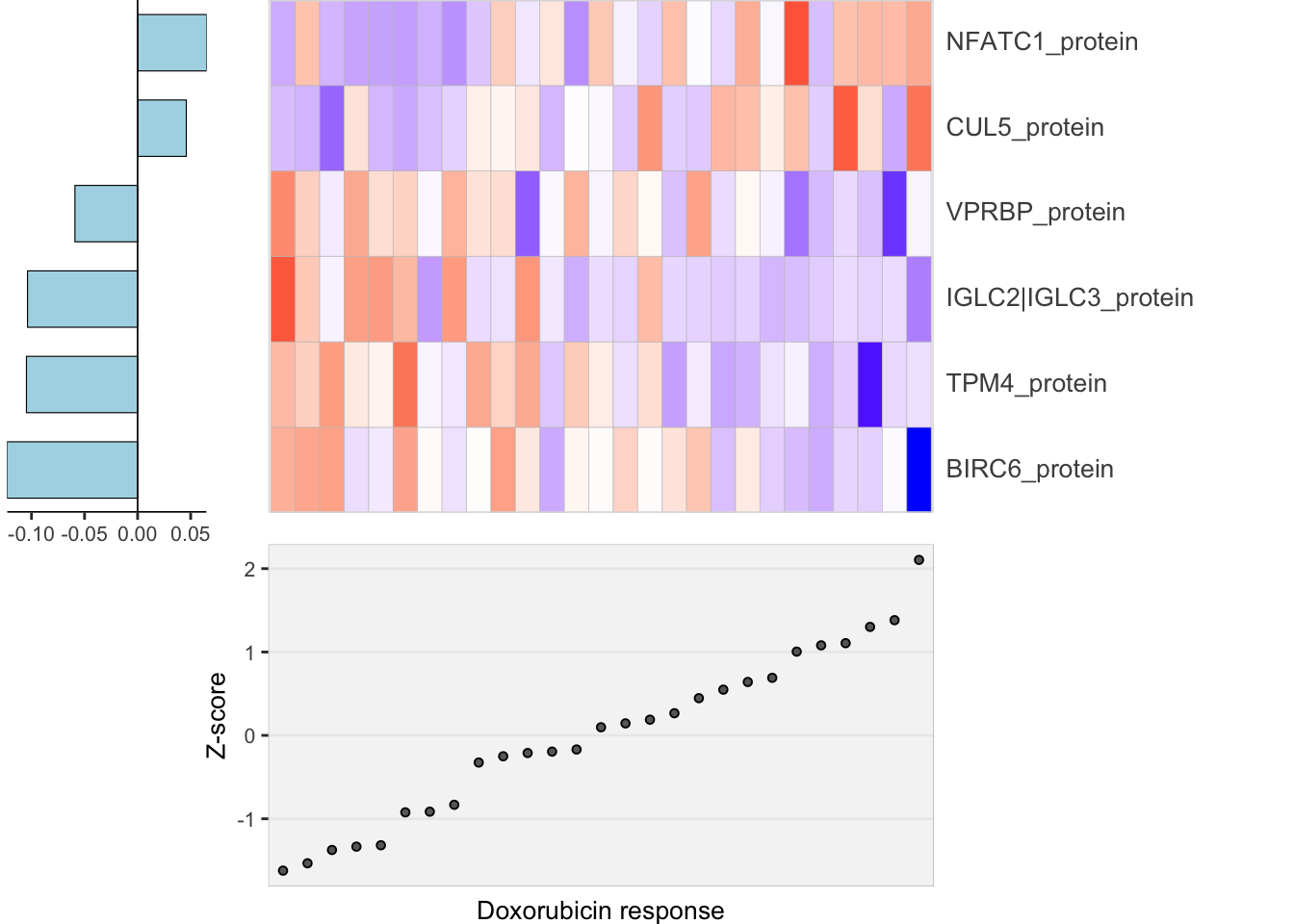
Only rna as predictors
Clean and integrate multi-omics data
inclSet<-list(RNA=exprMat)
cleanData <- generateData(responseList, inclSet, censor = 5)Run lasso
set.seed(2021)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
Variance explained
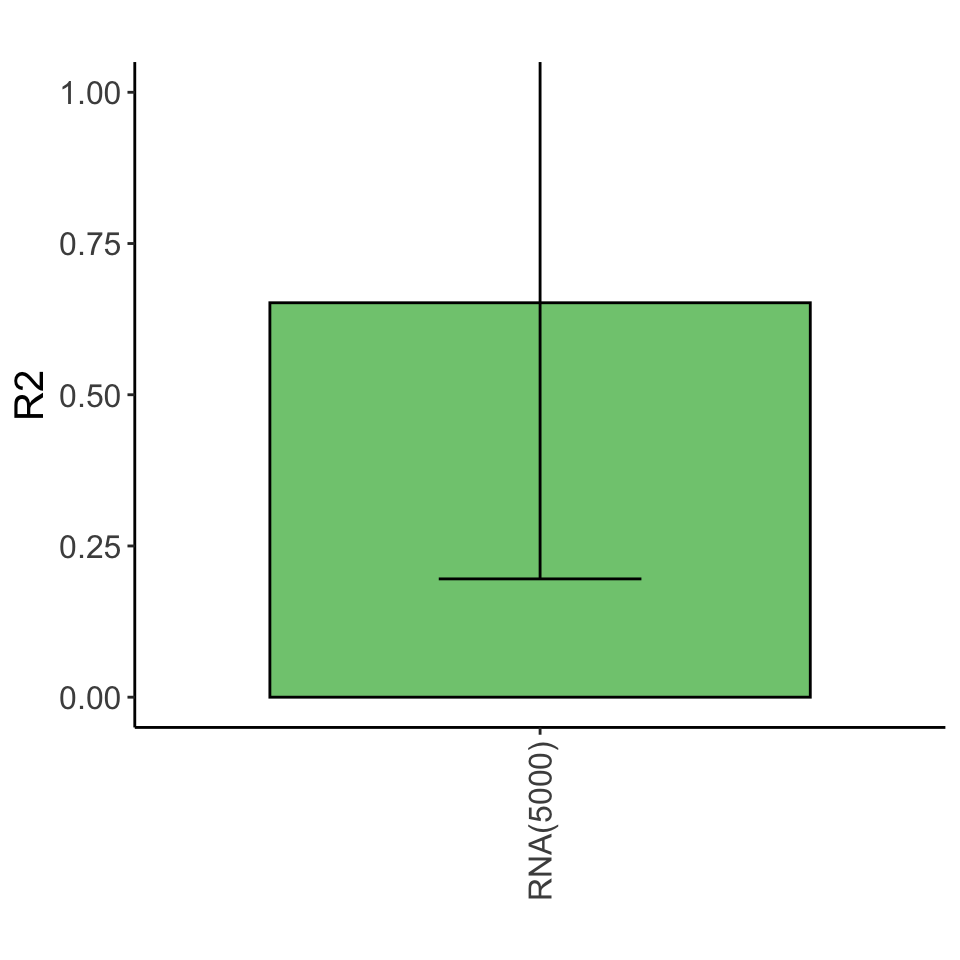
Only metabolites as predictors
Clean and integrate multi-omics data
inclSet<-list(Metabolite=metaMat)
cleanData <- generateData(responseList, inclSet, censor = 5)Run lasso
set.seed(2021)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
Variance explained
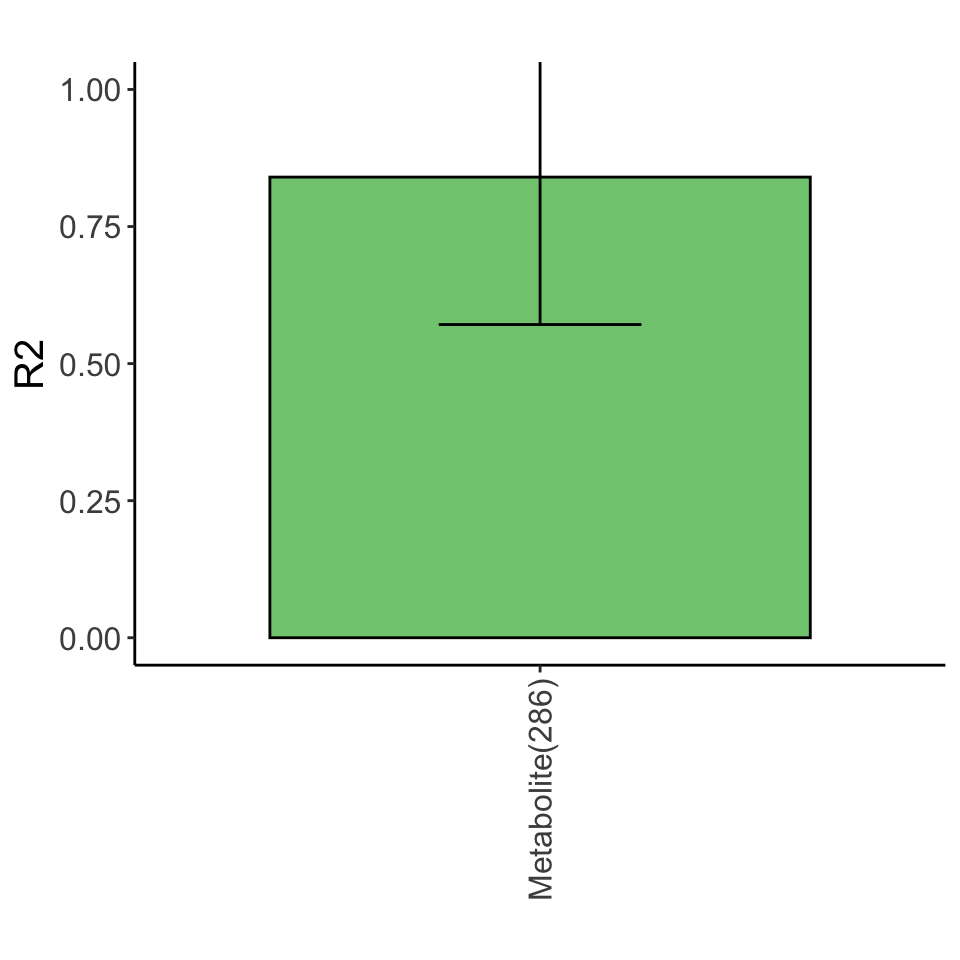
Lasso plot
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 0.8,setNumber = 1, legend = FALSE, scaleFac = 1)
heatMaps$Doxorubicin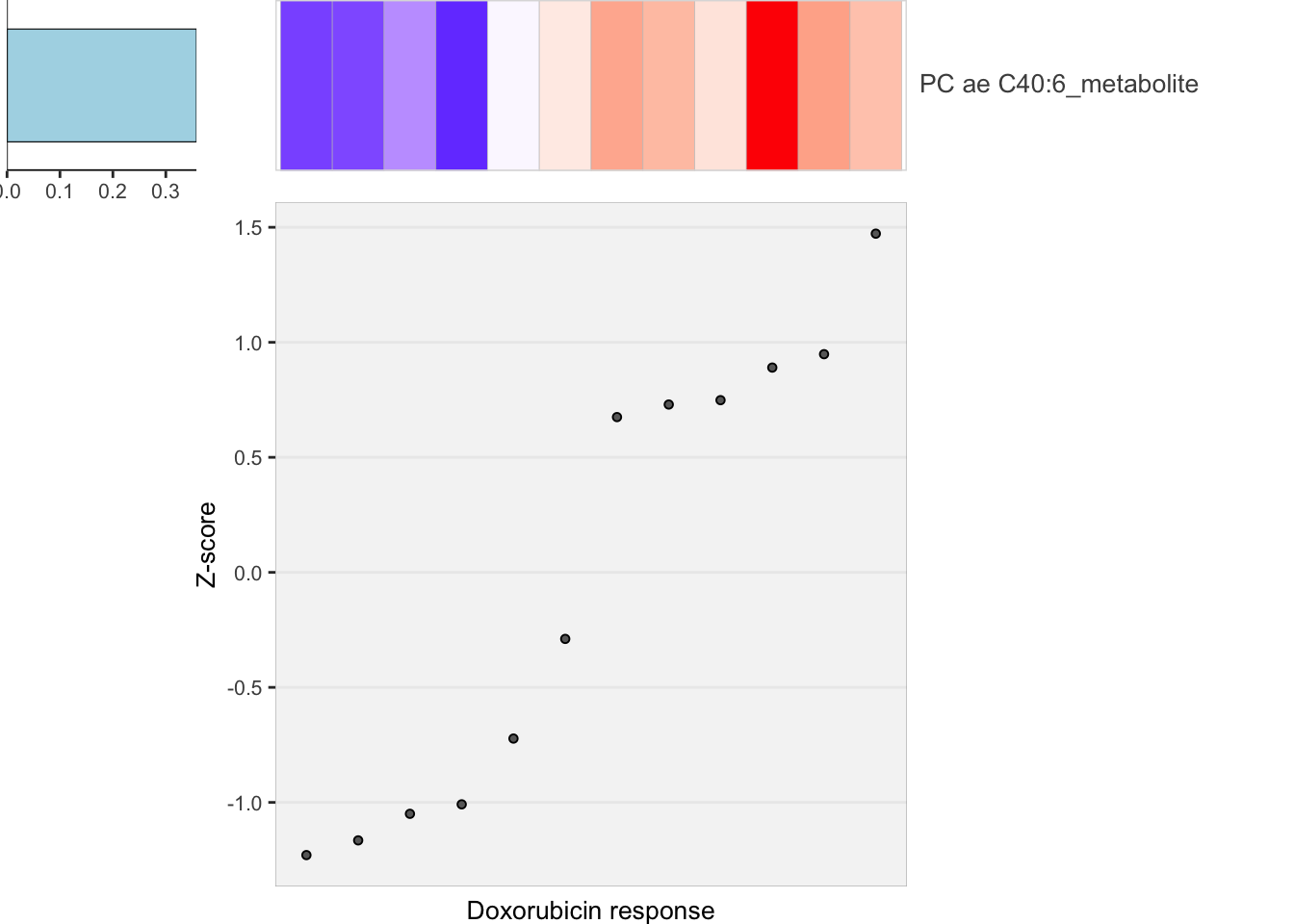
Heatmap of selected features
All combined
Clean and integrate multi-omics data
inclSet<-list(Gene=geneMat, Protein = protMat, RNA = exprMat, Metabolite = metaMat)
cleanData <- generateData(responseList, inclSet, censor = 5)set.seed(2021)
lassoResults <- list()
for (eachMeasure in names(cleanData$allResponse)) {
dataResult <- list()
for (eachDataset in names(cleanData$allExplain[[eachMeasure]])) {
y <- cleanData$allResponse[[eachMeasure]]
X <- cleanData$allExplain[[eachMeasure]][[eachDataset]]
glmRes <- runGlm(X, y, method = "lasso", repeats = 50, folds = 3)
dataResult[[eachDataset]] <- glmRes
}
lassoResults[[eachMeasure]] <- dataResult
}
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
varList <- plotVar(lassoResults)
cowplot::plot_grid(plotlist = varList, ncol=1)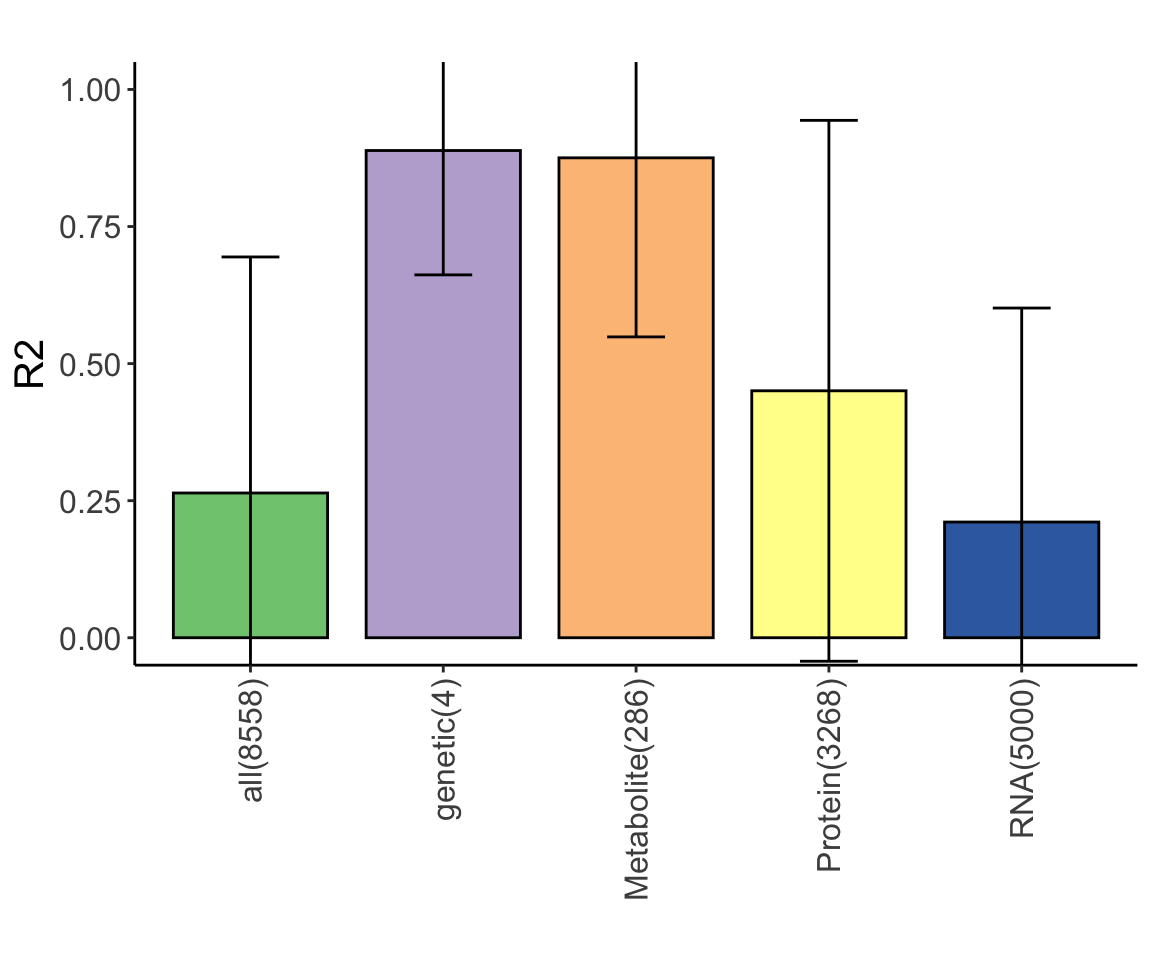
Combined
heatMaps <- lassoPlot(lassoResults, cleanData, freqCut = 0.2, setNumber = 5, legend = FALSE, scaleFac = 1)
plot_grid(heatMaps$Doxorubicin)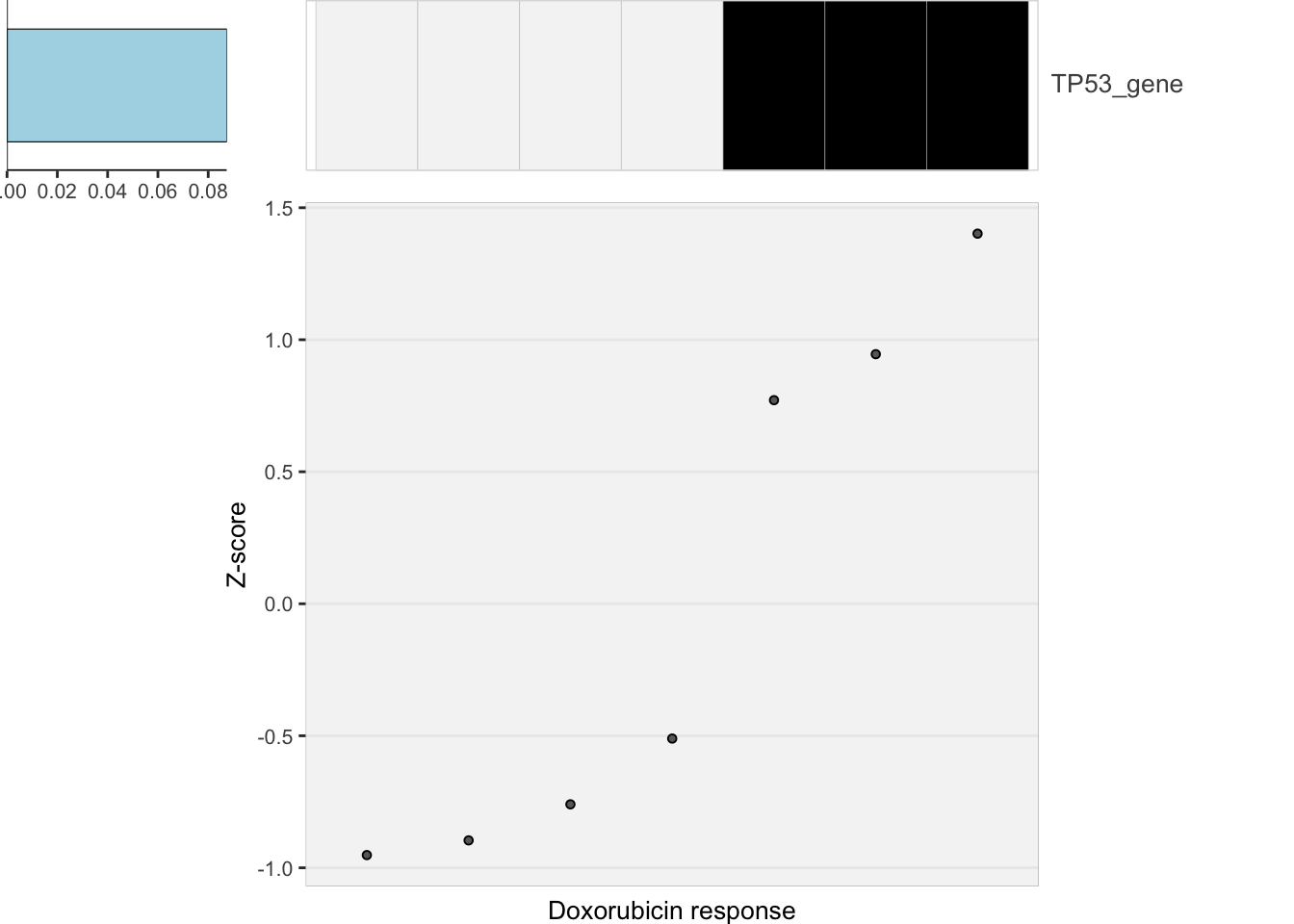 Sample size is perhaps too small for LASSO
Sample size is perhaps too small for LASSO
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] gtable_0.3.0 glmnet_4.1-4
[3] Matrix_1.4-1 pheatmap_1.0.12
[5] piano_2.12.0 forcats_0.5.1
[7] stringr_1.4.0 dplyr_1.0.9
[9] purrr_0.3.4 readr_2.1.2
[11] tidyr_1.2.0 tibble_3.1.7
[13] ggplot2_3.3.6 tidyverse_1.3.1
[15] proDA_1.10.0 cowplot_1.1.1
[17] DESeq2_1.36.0 SummarizedExperiment_1.26.1
[19] Biobase_2.56.0 GenomicRanges_1.48.0
[21] GenomeInfoDb_1.32.2 IRanges_2.30.0
[23] S4Vectors_0.34.0 BiocGenerics_0.42.0
[25] MatrixGenerics_1.8.0 matrixStats_0.62.0
[27] limma_3.52.2 jyluMisc_0.1.5
loaded via a namespace (and not attached):
[1] utf8_1.2.2 shinydashboard_0.7.2
[3] reticulate_1.25 tidyselect_1.1.2
[5] RSQLite_2.2.14 AnnotationDbi_1.58.0
[7] htmlwidgets_1.5.4 grid_4.2.0
[9] BiocParallel_1.30.3 Rtsne_0.16
[11] maxstat_0.7-25 munsell_0.5.0
[13] preprocessCore_1.58.0 codetools_0.2-18
[15] DT_0.23 withr_2.5.0
[17] colorspace_2.0-3 filelock_1.0.2
[19] highr_0.9 knitr_1.39
[21] rstudioapi_0.13 ggsignif_0.6.3
[23] labeling_0.4.2 git2r_0.30.1
[25] slam_0.1-50 GenomeInfoDbData_1.2.8
[27] KMsurv_0.1-5 bit64_4.0.5
[29] farver_2.1.0 rhdf5_2.40.0
[31] rprojroot_2.0.3 basilisk_1.8.0
[33] vctrs_0.4.1 generics_0.1.2
[35] TH.data_1.1-1 xfun_0.31
[37] sets_1.0-21 R6_2.5.1
[39] locfit_1.5-9.5 rhdf5filters_1.8.0
[41] bitops_1.0-7 cachem_1.0.6
[43] fgsea_1.22.0 DelayedArray_0.22.0
[45] assertthat_0.2.1 promises_1.2.0.1
[47] scales_1.2.0 multcomp_1.4-19
[49] extraDistr_1.9.1 egg_0.4.5
[51] affy_1.74.0 sandwich_3.0-2
[53] workflowr_1.7.0 rlang_1.0.2
[55] genefilter_1.78.0 splines_4.2.0
[57] rstatix_0.7.0 broom_0.8.0
[59] reshape2_1.4.4 BiocManager_1.30.18
[61] yaml_2.3.5 abind_1.4-5
[63] modelr_0.1.8 crosstalk_1.2.0
[65] backports_1.4.1 httpuv_1.6.5
[67] tools_4.2.0 relations_0.6-12
[69] affyio_1.66.0 ellipsis_0.3.2
[71] gplots_3.1.3 jquerylib_0.1.4
[73] RColorBrewer_1.1-3 MultiAssayExperiment_1.22.0
[75] plyr_1.8.7 Rcpp_1.0.8.3
[77] visNetwork_2.1.0 zlibbioc_1.42.0
[79] RCurl_1.98-1.7 basilisk.utils_1.8.0
[81] ggpubr_0.4.0 zoo_1.8-10
[83] haven_2.5.0 ggrepel_0.9.1
[85] cluster_2.1.3 exactRankTests_0.8-35
[87] fs_1.5.2 magrittr_2.0.3
[89] data.table_1.14.2 reprex_2.0.1
[91] survminer_0.4.9 mvtnorm_1.1-3
[93] hms_1.1.1 shinyjs_2.1.0
[95] mime_0.12 evaluate_0.15
[97] xtable_1.8-4 XML_3.99-0.10
[99] readxl_1.4.0 shape_1.4.6
[101] gridExtra_2.3 compiler_4.2.0
[103] KernSmooth_2.23-20 crayon_1.5.1
[105] htmltools_0.5.2 mgcv_1.8-40
[107] later_1.3.0 tzdb_0.3.0
[109] geneplotter_1.74.0 lubridate_1.8.0
[111] DBI_1.1.3 corrplot_0.92
[113] dbplyr_2.2.0 MASS_7.3-57
[115] car_3.1-0 cli_3.3.0
[117] vsn_3.64.0 marray_1.74.0
[119] parallel_4.2.0 igraph_1.3.2
[121] pkgconfig_2.0.3 km.ci_0.5-6
[123] dir.expiry_1.4.0 foreach_1.5.2
[125] xml2_1.3.3 annotate_1.74.0
[127] bslib_0.3.1 XVector_0.36.0
[129] drc_3.0-1 rvest_1.0.2
[131] digest_0.6.29 Biostrings_2.64.0
[133] rmarkdown_2.14 cellranger_1.1.0
[135] fastmatch_1.1-3 survMisc_0.5.6
[137] uwot_0.1.11 shiny_1.7.1
[139] gtools_3.9.2.2 lifecycle_1.0.1
[141] nlme_3.1-158 jsonlite_1.8.0
[143] Rhdf5lib_1.18.2 carData_3.0-5
[145] fansi_1.0.3 pillar_1.7.0
[147] lattice_0.20-45 KEGGREST_1.36.2
[149] fastmap_1.1.0 httr_1.4.3
[151] plotrix_3.8-2 survival_3.3-1
[153] glue_1.6.2 iterators_1.0.14
[155] png_0.1-7 bit_4.0.4
[157] HDF5Array_1.24.1 stringi_1.7.6
[159] sass_0.4.1 blob_1.2.3
[161] caTools_1.18.2 memoise_2.0.1