Integrative analysis using MOFA
Junyan Lu
2022-10-20
Last updated: 2022-11-10
Checks: 5 1
Knit directory: irAE_LungCancer/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load packages
library(MultiAssayExperiment)
library(MOFA2)
library(jyluMisc)
library(tidyverse)
knitr::opts_chunk$set(warning = FALSE, message = FALSE)Preprocessing datsets
Load data
load("../output/processedData.RData")#mae <- mae[,mae$condition!="noMalignancy"]
colData(mae) <- colData(mae)[,c("patID","condition","Group")]CBA data
cbaMat <- mae[["cba"]]
cbaMat <- glog2(cbaMat)
mae[["cba"]] <- cbaMatNMR data
nmrMat <- mae[["nmr"]]Create a new group with follow_up - baseline
CBA
cbaBase <- glog2(mae[,mae$condition == "Baseline"][["cba"]])
colnames(cbaBase) <- mae[,mae$condition == "Baseline"]$patID
cbaFollow <- glog2(mae[,mae$condition == "Follow_Up"][["cba"]])
colnames(cbaFollow) <- mae[,mae$condition == "Follow_Up"]$patID
allPat <- unique(c(colnames(cbaBase), colnames(cbaFollow)))
cbaDiff <- cbaFollow[,match(allPat,colnames(cbaFollow))] - cbaBase[,match(allPat,colnames(cbaBase))]
colnames(cbaDiff) <- allPat
cbaDiff <- cbaDiff[,complete.cases(t(cbaDiff))]NMR
nmrBase <- mae[,mae$condition == "Baseline"][["nmr"]]
colnames(nmrBase) <- mae[,mae$condition == "Baseline"]$patID
nmrFollow <- mae[,mae$condition == "Follow_Up"][["nmr"]]
colnames(nmrFollow) <- mae[,mae$condition == "Follow_Up"]$patID
allPat <- unique(c(colnames(nmrBase), colnames(nmrFollow)))
nmrDiff <- nmrFollow[,match(allPat,colnames(nmrFollow))] - nmrBase[,match(allPat,colnames(nmrBase))]
colnames(nmrDiff) <- allPat
nmrDiff <- nmrDiff[,complete.cases(t(nmrDiff))]Combine and create new mae object
matList <- list(cba = cbind(cbaMat, cbaDiff),
nmr = cbind(nmrMat, nmrDiff))
#create new annotation
diffPatAnno <- colData(mae[,mae$condition != "noMalignancy"])
diffPatAnno <- diffPatAnno[!duplicated(diffPatAnno$patID),]
diffPatAnno$condition <- "Follow_Up_Baseline_diff"
rownames(diffPatAnno) <- diffPatAnno$patID
diffPatAnno <- diffPatAnno[rownames(diffPatAnno) %in% c(colnames(cbaDiff), colnames(nmrDiff)),]
newColData <- rbind(colData(mae), diffPatAnno)[,c("patID","condition","Group")]
maeNew <- MultiAssayExperiment(matList, colData = newColData)Create and prepare MOFA object
Create MOFA object
MOFAobject <- create_mofa(maeNew, groups = "condition")Plot data overview
plot_data_overview(MOFAobject)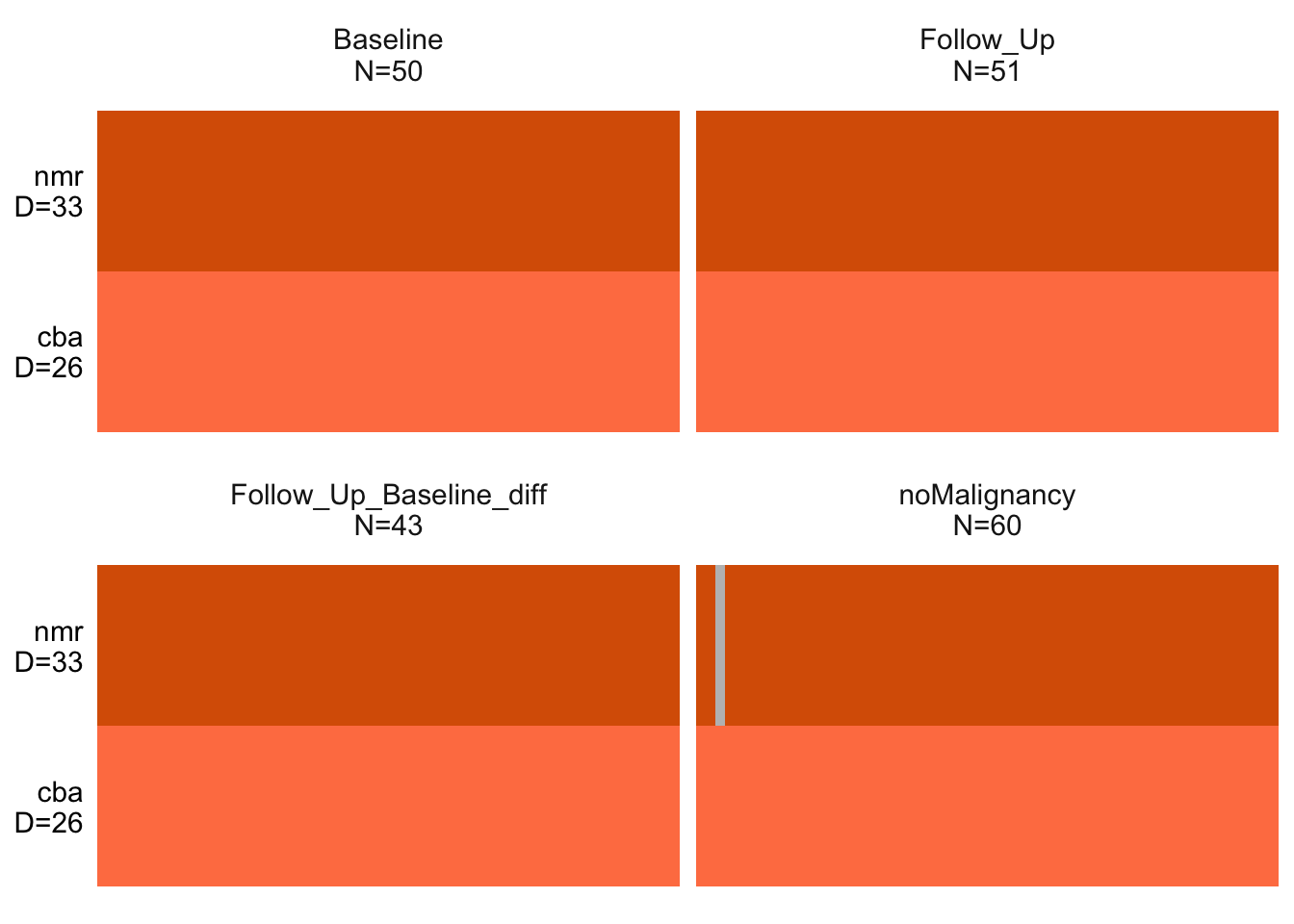
Data options
data_opts <- get_default_data_options(MOFAobject)
data_opts$scale_views=TRUE
data_opts$scale_views
[1] TRUE
$scale_groups
[1] FALSE
$center_groups
[1] TRUE
$use_float32
[1] FALSE
$views
[1] "cba" "nmr"
$groups
[1] "Baseline" "Follow_Up"
[3] "Follow_Up_Baseline_diff" "noMalignancy" Model options
model_opts <- get_default_model_options(MOFAobject)
model_opts$num_factors <- 30
model_opts$likelihoods
cba nmr
"gaussian" "gaussian"
$num_factors
[1] 30
$spikeslab_factors
[1] FALSE
$spikeslab_weights
[1] TRUE
$ard_factors
[1] TRUE
$ard_weights
[1] TRUETraining options
train_opts <- get_default_training_options(MOFAobject)
train_opts$convergence_mode <- "slow"
train_opts$seed <- 2022
train_opts$maxiter <- 10000
train_opts$maxiter
[1] 10000
$convergence_mode
[1] "slow"
$drop_factor_threshold
[1] -1
$verbose
[1] FALSE
$startELBO
[1] 1
$freqELBO
[1] 5
$stochastic
[1] FALSE
$gpu_mode
[1] FALSE
$seed
[1] 2022
$outfile
NULL
$weight_views
[1] FALSE
$save_interrupted
[1] FALSEChange drop threshold to 0.01
train_opts$drop_factor_threshold <-0.01Train the MOFA model
Prepare MOFA object
MOFAobject <- prepare_mofa(MOFAobject,
data_options = data_opts,
model_options = model_opts,
training_options = train_opts
)Training
MOFAobject <- run_mofa(MOFAobject )
save(MOFAobject, file= "../output/mofaRes.RData")Load trained model
load("../output/mofaRes.RData")Preliminary analysis of the results
Factor correlation matrix
plot_factor_cor(MOFAobject)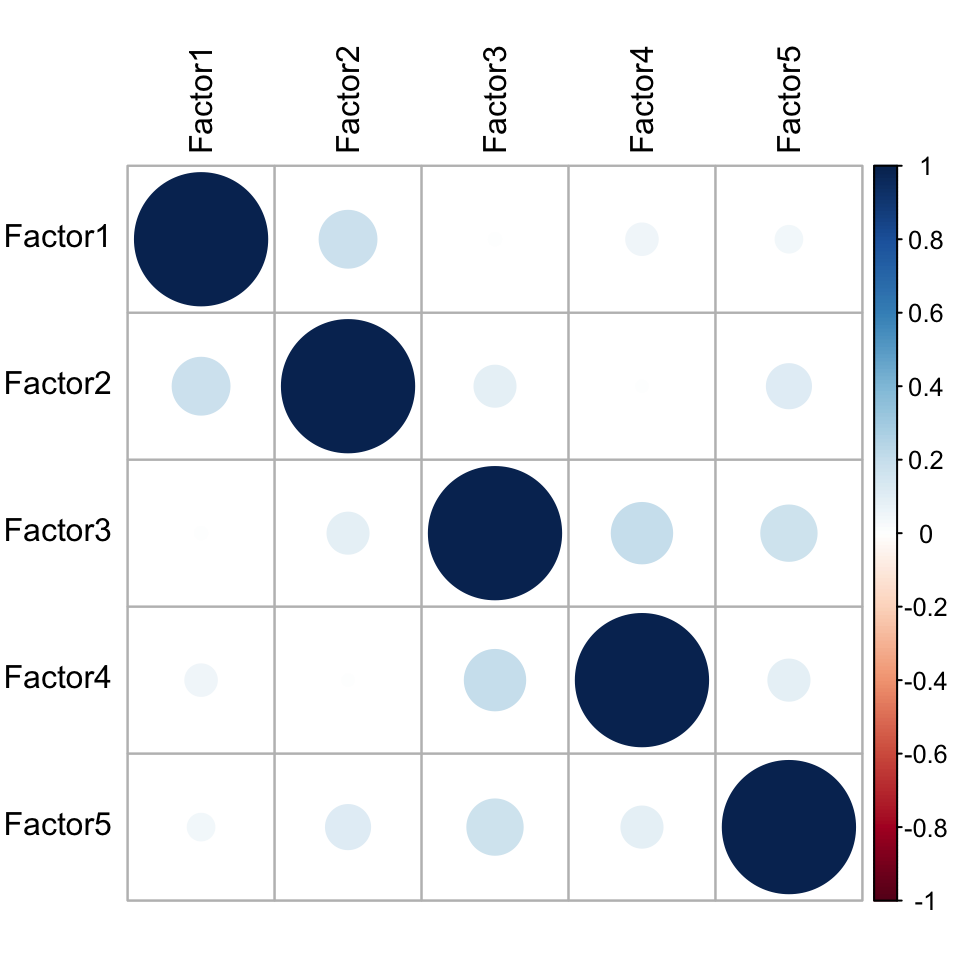
Variance explained
plot_variance_explained(MOFAobject, max_r2=15)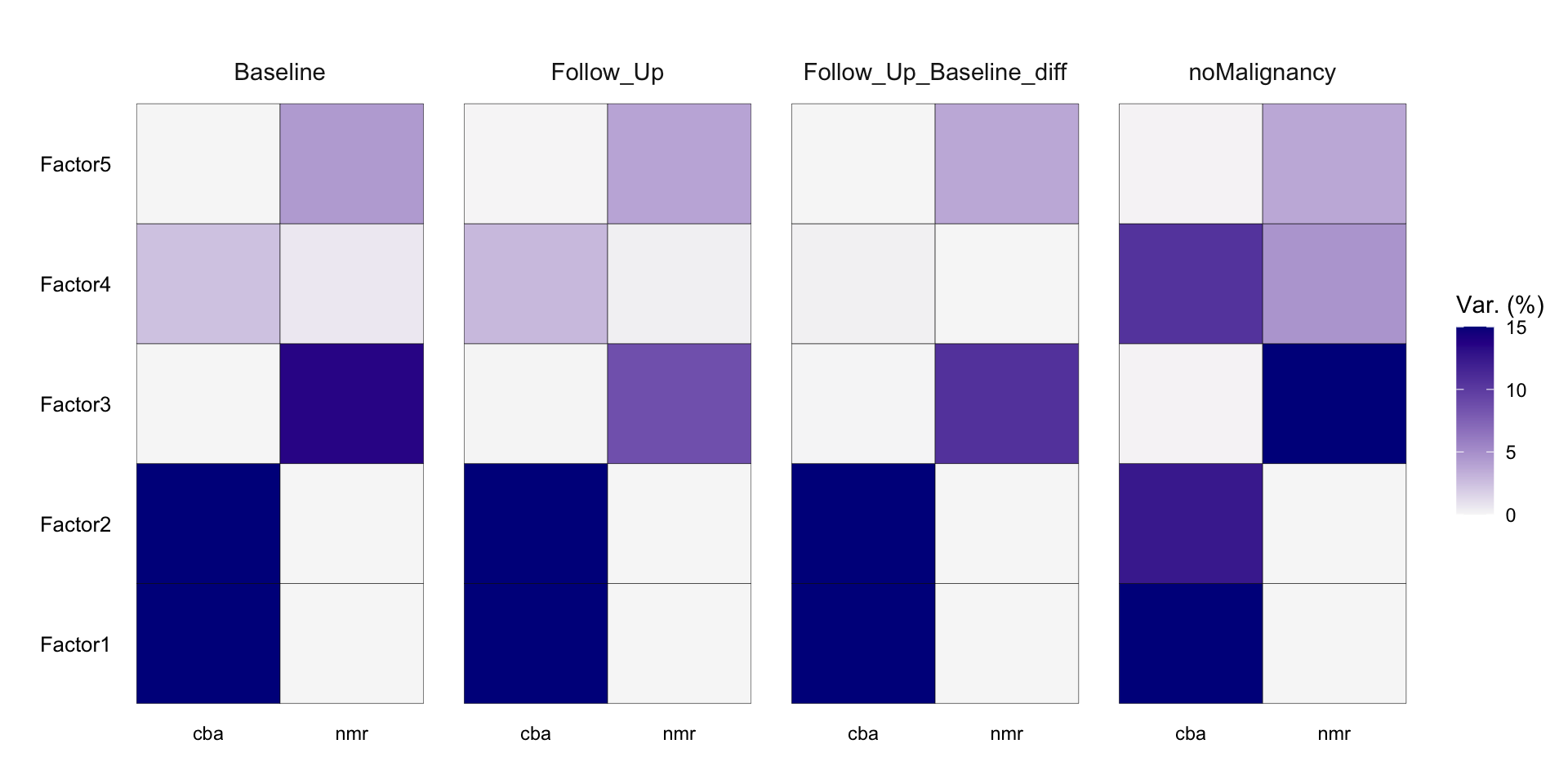
Total variance explained
plot_variance_explained(MOFAobject, plot_total = T)[[2]]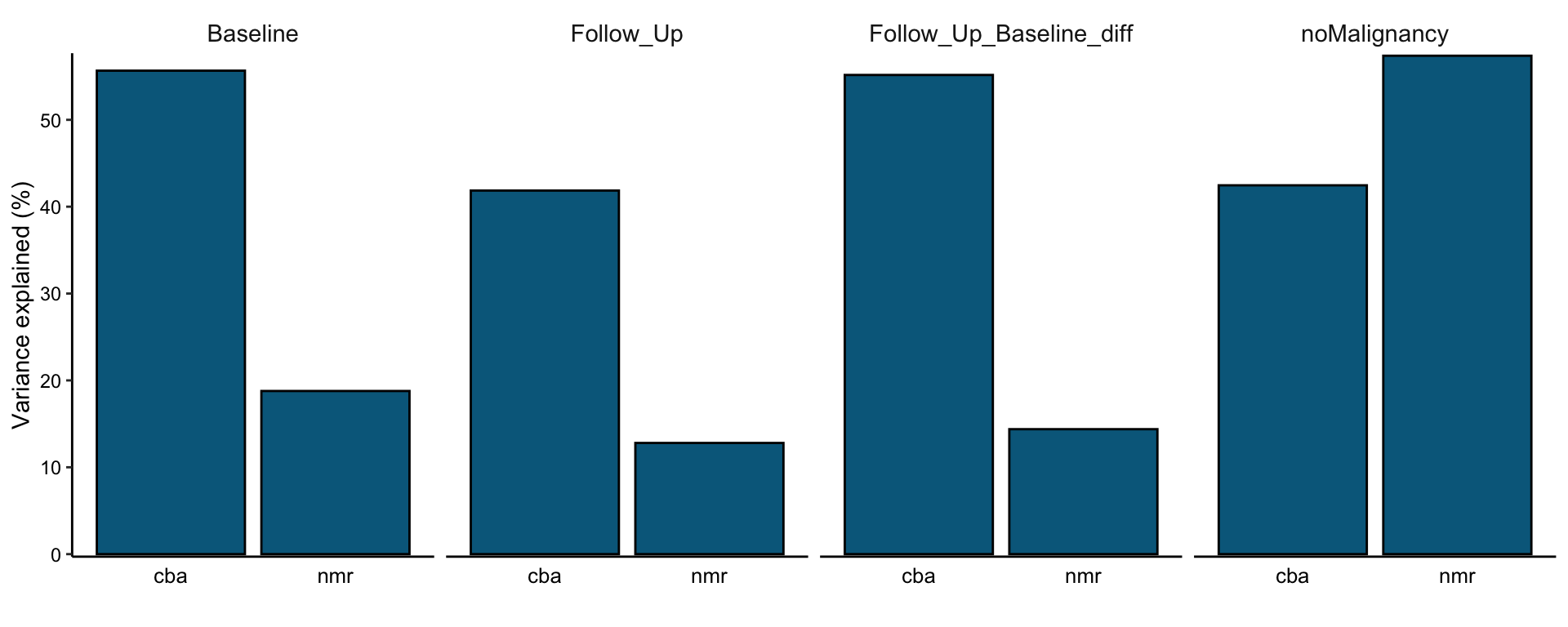
Associate factor with Group
allFact <- get_factors(MOFAobject)
facTab <- lapply(names(allFact), function(x) {
allFact[[x]] %>% as_tibble(rownames = "sampleID") %>%
pivot_longer(-sampleID, names_to = "factor", values_to = "value") %>%
mutate(mofaGroup = x)
}) %>% bind_rows
patAnno <- colData(maeNew) %>% as_tibble(rownames = "sampleID")
facTab <- left_join(facTab, patAnno, by = "sampleID")resTab <- group_by(facTab, factor, condition) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group, .)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% arrange(p.value) %>%
filter(term == "Group") %>%
select(factor, condition, p.value)
resTab.sig <- filter(resTab, p.value < 0.1)
resTab.sig# A tibble: 7 × 3
# Groups: factor, condition [7]
factor condition p.value
<chr> <chr> <dbl>
1 Factor3 noMalignancy 0.0000101
2 Factor2 noMalignancy 0.000117
3 Factor2 Follow_Up 0.000547
4 Factor4 noMalignancy 0.00508
5 Factor2 Follow_Up_Baseline_diff 0.00580
6 Factor5 noMalignancy 0.0105
7 Factor4 Follow_Up 0.0201 Plot associations
pList <- lapply(seq(nrow(resTab.sig)), function(i) {
rec <- resTab.sig[i,]
plotTab <- filter(facTab, factor == rec$factor, condition == rec$condition)
ggplot(plotTab, aes(x=Group, y=value)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = Group)) +
theme_bw() +
ggtitle(sprintf("%s (%s)", rec$factor, rec$condition))
})
cowplot::plot_grid(plotlist = pList, ncol=3)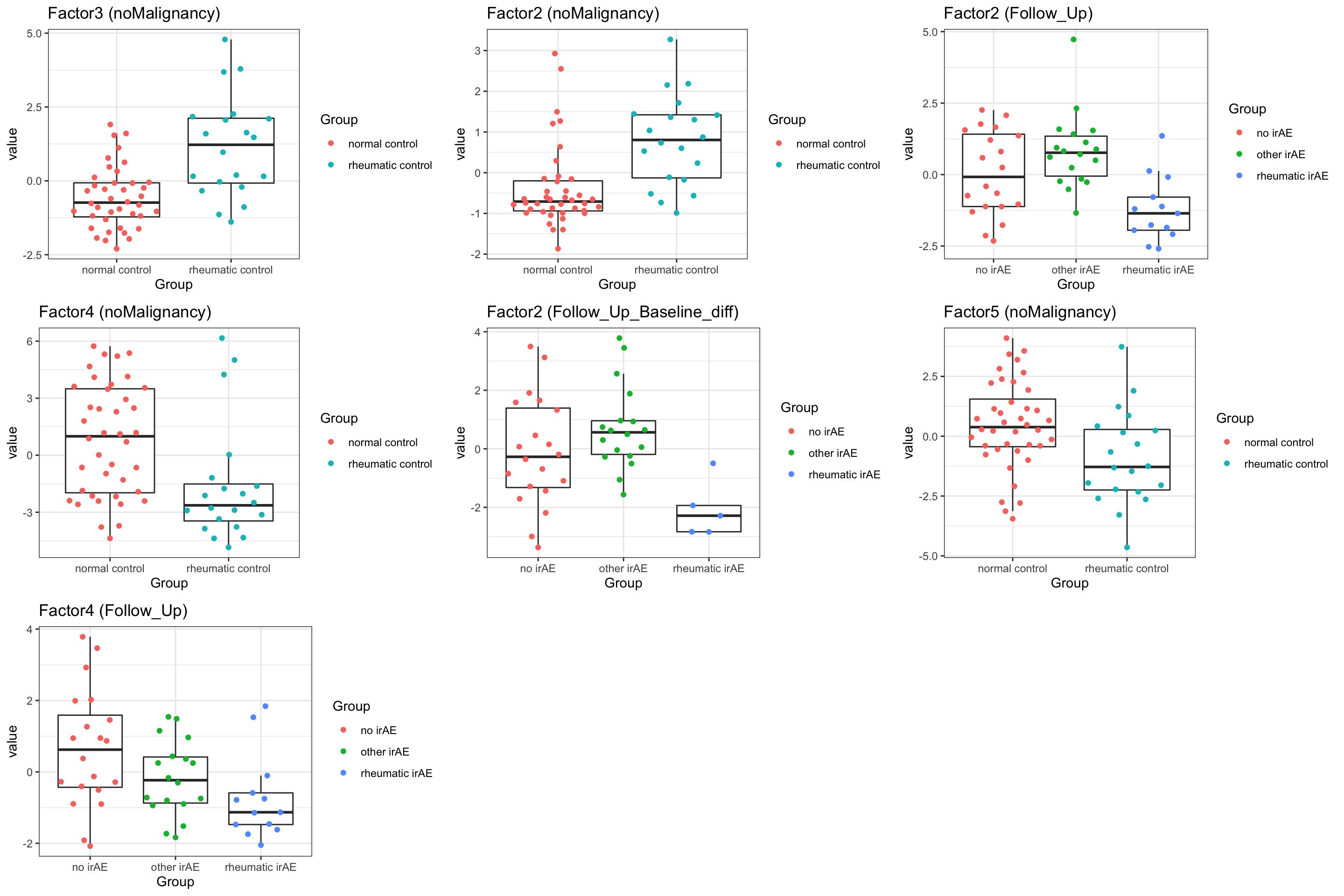
Factor heatmap
facGroupTab <- facTab %>% mutate(facGroup = paste0(factor, "_", mofaGroup)) %>%
filter(facGroup %in% paste0(resTab.sig$factor, "_", resTab.sig$condition)) %>%
filter(condition != "noMalignancy")
facMat <- facGroupTab %>% select(facGroup, patID, value) %>%
pivot_wider(names_from = patID, values_from = value) %>%
column_to_rownames("facGroup") %>% as.matrix()
colAnno <- facGroupTab %>% distinct(patID, Group) %>%
column_to_rownames("patID") %>% data.frame()
pheatmap::pheatmap(facMat, annotation_col = colAnno, clustering_method = "ward.D2", scale ="row")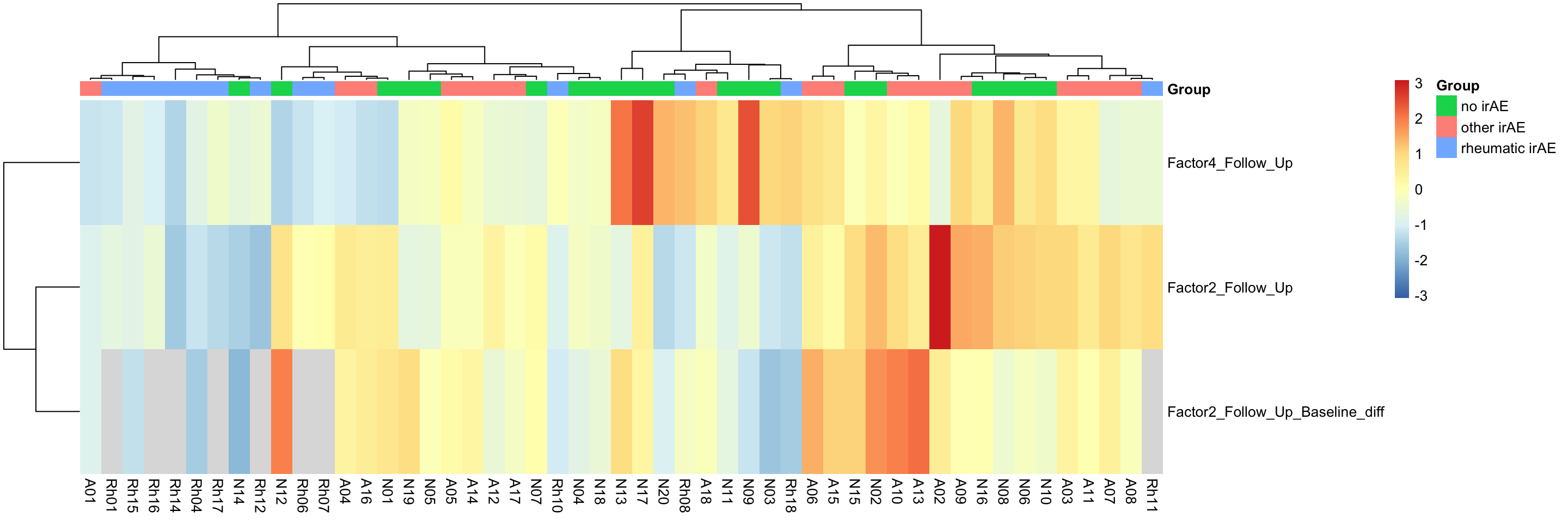
Scatter plots
Factor 2 Follow_up versus Factor 4 Follow up
plotTab <- filter(facGroupTab, facGroup %in% c("Factor2_Follow_Up", "Factor4_Follow_Up")) %>%
select(patID, Group, facGroup, value) %>%
pivot_wider(names_from = facGroup, values_from = value)
ggplot(plotTab, aes(x= Factor2_Follow_Up, y=Factor4_Follow_Up)) +
geom_point(aes(col = Group)) +
theme_bw()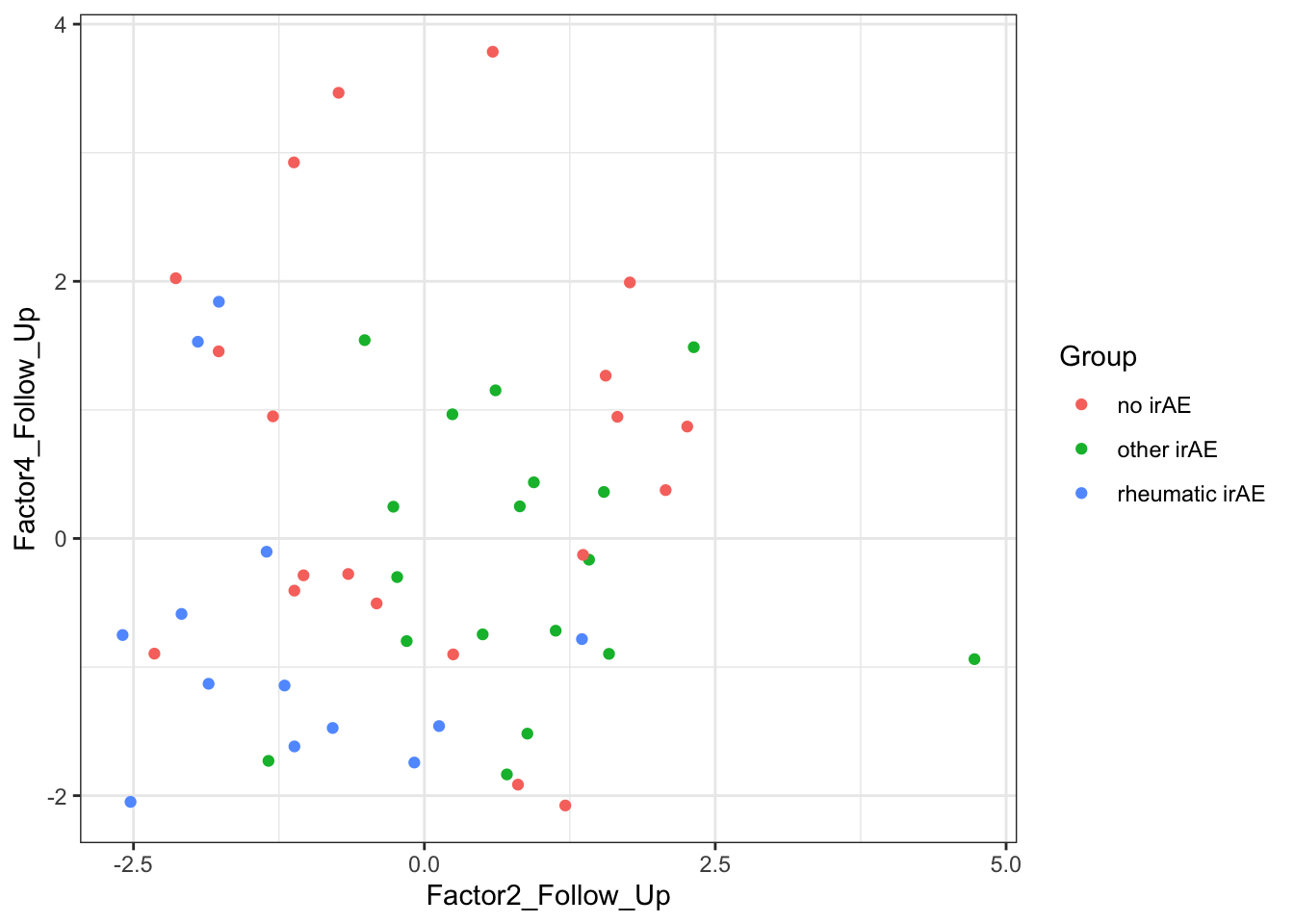
Factor loadings
Factor 2
CBA
plot_weights(MOFAobject, view = "cba", factors = 2)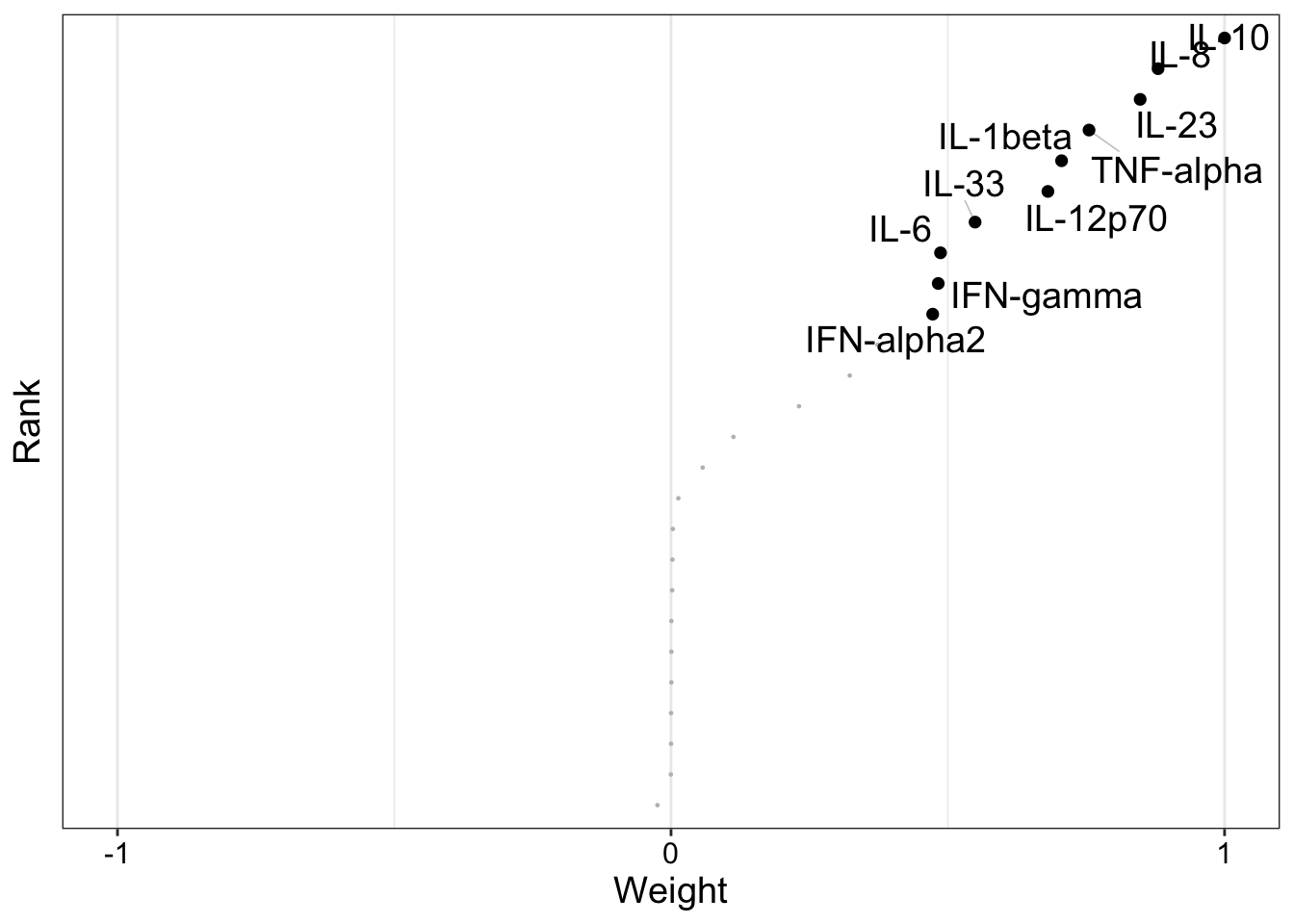
NMR
plot_weights(MOFAobject, view = "nmr", factors = 2)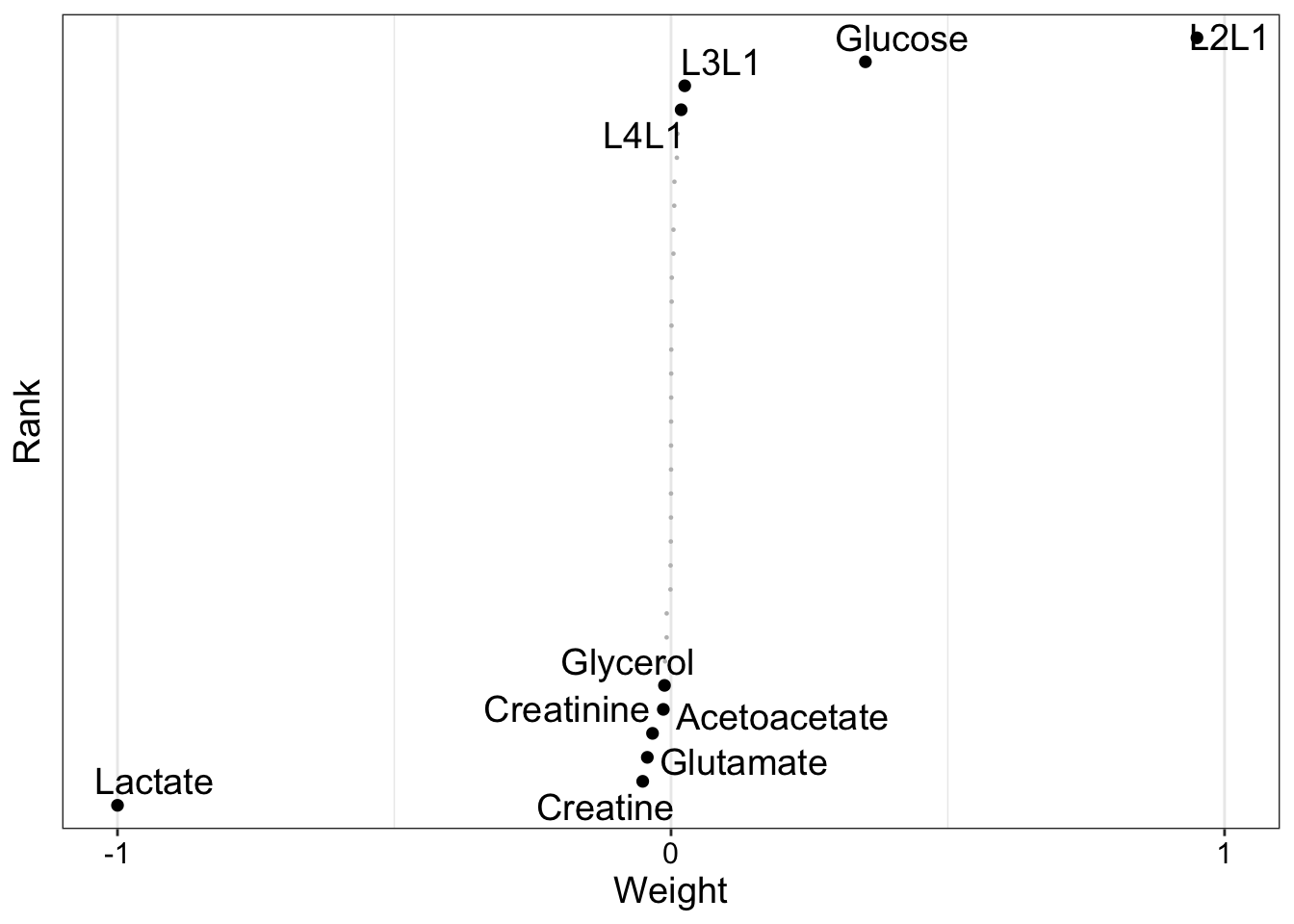
Factor 4
CBA
plot_weights(MOFAobject, view = "cba", factors = 4)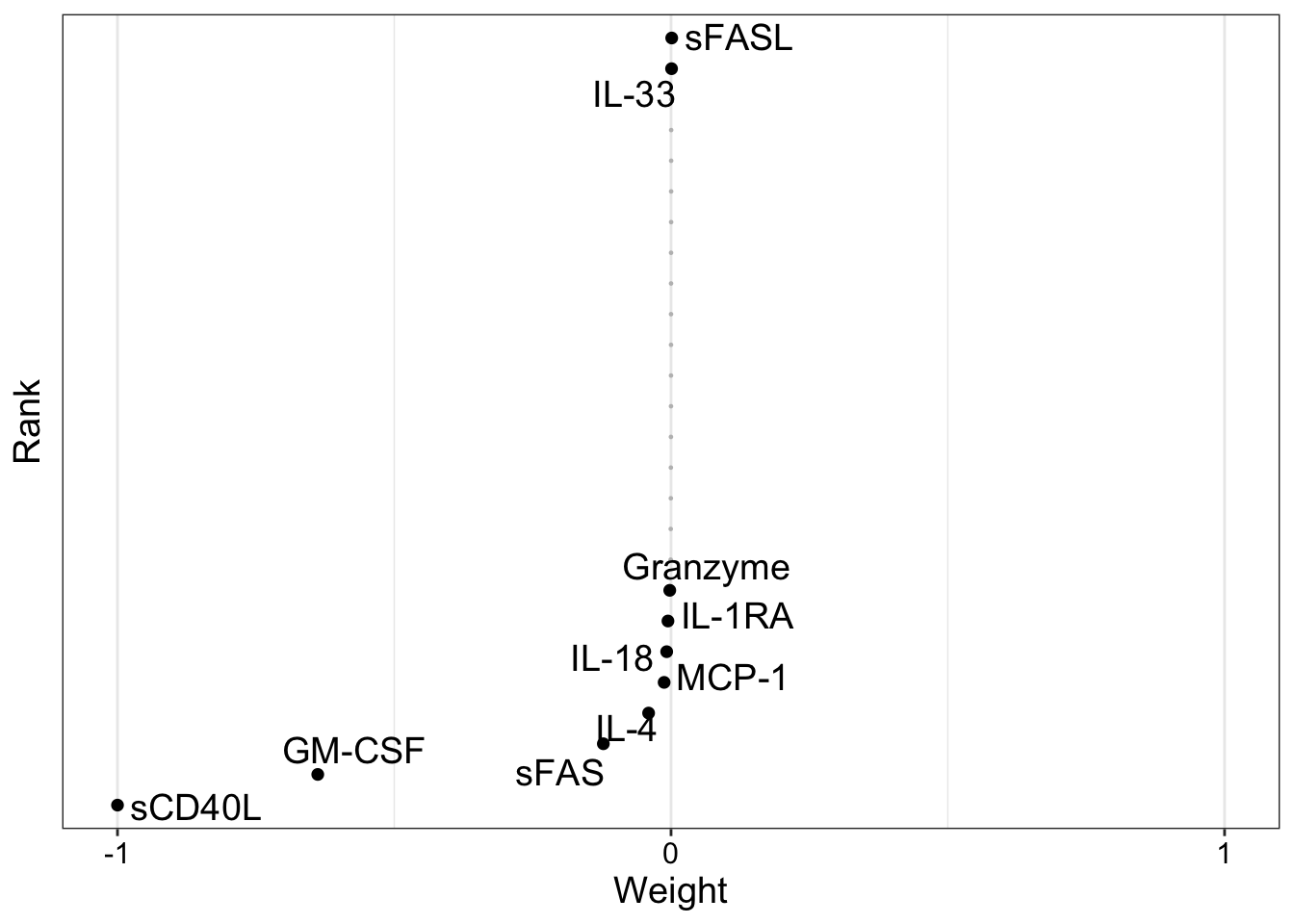
NMR
plot_weights(MOFAobject, view = "nmr", factors = 4)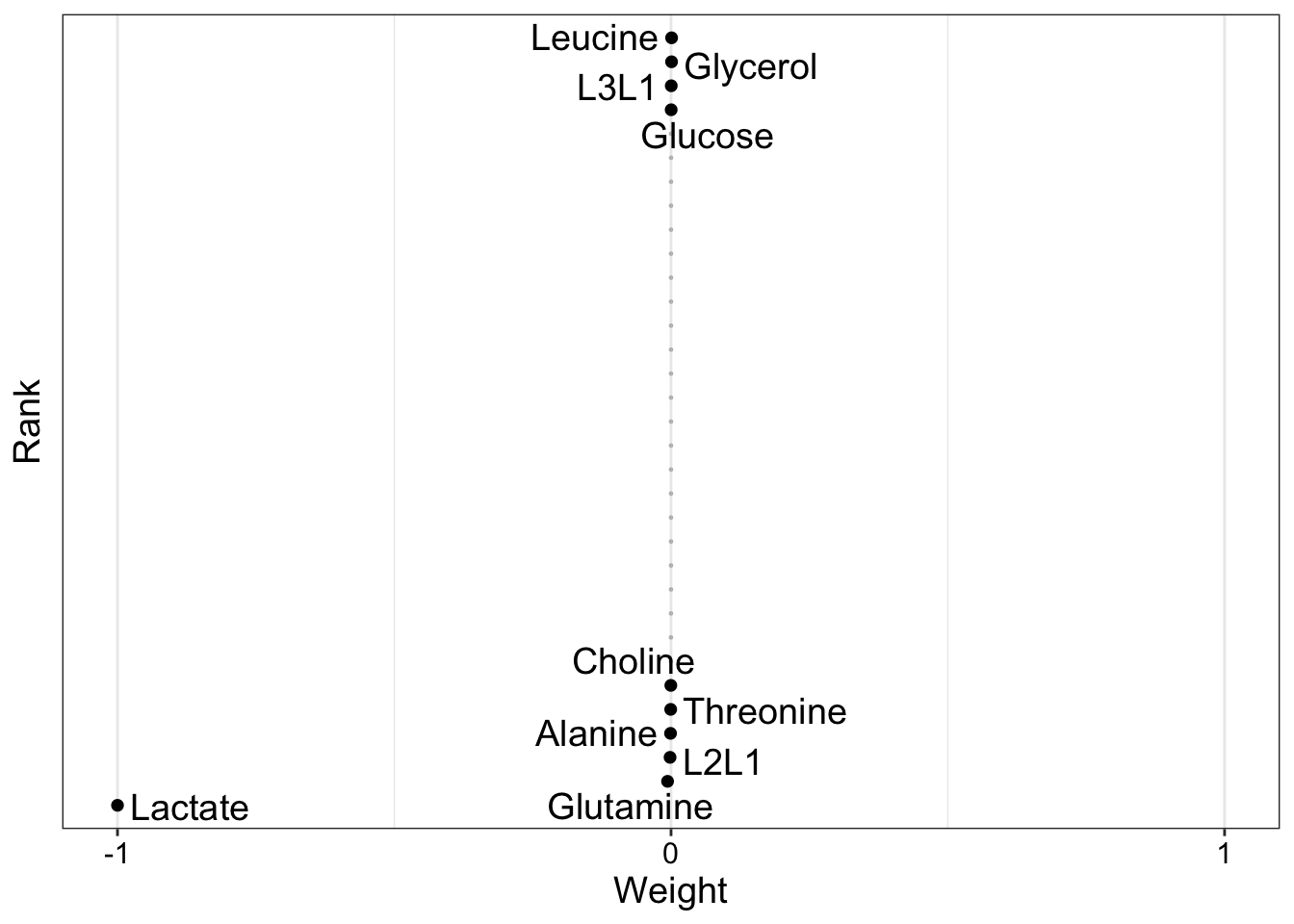
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.0
[3] dplyr_1.0.9 purrr_0.3.4
[5] readr_2.1.2 tidyr_1.2.0
[7] tibble_3.1.8 ggplot2_3.3.6
[9] tidyverse_1.3.2 jyluMisc_0.1.5
[11] MOFA2_1.6.0 MultiAssayExperiment_1.22.0
[13] SummarizedExperiment_1.26.1 Biobase_2.56.0
[15] GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[17] IRanges_2.30.0 S4Vectors_0.34.0
[19] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[21] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] readxl_1.4.0 backports_1.4.1 fastmatch_1.1-3
[4] drc_3.0-1 corrplot_0.92 workflowr_1.7.0
[7] plyr_1.8.7 igraph_1.3.4 shinydashboard_0.7.2
[10] splines_4.2.0 BiocParallel_1.30.3 TH.data_1.1-1
[13] digest_0.6.29 htmltools_0.5.3 fansi_1.0.3
[16] magrittr_2.0.3 googlesheets4_1.0.0 cluster_2.1.3
[19] tzdb_0.3.0 limma_3.52.2 modelr_0.1.8
[22] sandwich_3.0-2 piano_2.12.0 colorspace_2.0-3
[25] rvest_1.0.2 ggrepel_0.9.1 haven_2.5.0
[28] xfun_0.31 crayon_1.5.1 RCurl_1.98-1.7
[31] jsonlite_1.8.0 survival_3.4-0 zoo_1.8-10
[34] glue_1.6.2 survminer_0.4.9 gargle_1.2.0
[37] gtable_0.3.0 zlibbioc_1.42.0 XVector_0.36.0
[40] DelayedArray_0.22.0 car_3.1-0 Rhdf5lib_1.18.2
[43] HDF5Array_1.24.1 abind_1.4-5 scales_1.2.0
[46] pheatmap_1.0.12 mvtnorm_1.1-3 DBI_1.1.3
[49] relations_0.6-12 rstatix_0.7.0 Rcpp_1.0.9
[52] plotrix_3.8-2 xtable_1.8-4 reticulate_1.25
[55] km.ci_0.5-6 DT_0.23 httr_1.4.3
[58] htmlwidgets_1.5.4 fgsea_1.22.0 dir.expiry_1.4.0
[61] gplots_3.1.3 RColorBrewer_1.1-3 ellipsis_0.3.2
[64] farver_2.1.1 pkgconfig_2.0.3 dbplyr_2.2.1
[67] sass_0.4.2 uwot_0.1.11 utf8_1.2.2
[70] labeling_0.4.2 tidyselect_1.1.2 rlang_1.0.4
[73] reshape2_1.4.4 later_1.3.0 cellranger_1.1.0
[76] munsell_0.5.0 tools_4.2.0 visNetwork_2.1.0
[79] cachem_1.0.6 cli_3.3.0 generics_0.1.3
[82] broom_1.0.0 evaluate_0.15 fastmap_1.1.0
[85] yaml_2.3.5 knitr_1.39 fs_1.5.2
[88] survMisc_0.5.6 caTools_1.18.2 mime_0.12
[91] slam_0.1-50 xml2_1.3.3 compiler_4.2.0
[94] rstudioapi_0.13 beeswarm_0.4.0 filelock_1.0.2
[97] png_0.1-7 ggsignif_0.6.3 marray_1.74.0
[100] reprex_2.0.1 bslib_0.4.0 stringi_1.7.8
[103] highr_0.9 basilisk.utils_1.8.0 lattice_0.20-45
[106] Matrix_1.4-1 shinyjs_2.1.0 KMsurv_0.1-5
[109] vctrs_0.4.1 pillar_1.8.0 lifecycle_1.0.1
[112] rhdf5filters_1.8.0 jquerylib_0.1.4 data.table_1.14.2
[115] cowplot_1.1.1 bitops_1.0-7 httpuv_1.6.5
[118] R6_2.5.1 promises_1.2.0.1 KernSmooth_2.23-20
[121] gridExtra_2.3 vipor_0.4.5 codetools_0.2-18
[124] MASS_7.3-58 gtools_3.9.3 exactRankTests_0.8-35
[127] assertthat_0.2.1 rhdf5_2.40.0 rprojroot_2.0.3
[130] withr_2.5.0 multcomp_1.4-19 GenomeInfoDbData_1.2.8
[133] hms_1.1.1 parallel_4.2.0 grid_4.2.0
[136] basilisk_1.8.0 rmarkdown_2.14 googledrive_2.0.0
[139] carData_3.0-5 Rtsne_0.16 git2r_0.30.1
[142] maxstat_0.7-25 ggpubr_0.4.0 sets_1.0-21
[145] lubridate_1.8.0 shiny_1.7.2 ggbeeswarm_0.6.0