Perform association tests between molecular features and disease phenotypes for each dataset
Junyan Lu
Last updated: 2022-11-11
Checks: 5 1
Knit directory: irAE_LungCancer/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load packages
library(MultiAssayExperiment)
library(pheatmap)
library(vsn)
library(jyluMisc)
library(tidyverse)
knitr::opts_chunk$set(warning = FALSE, message = FALSE)Load data
load("../output/processedData.RData")
patAnno <- colData(mae) %>%
as_tibble(rownames = "sampleID")Analyses focused on cancer patients
CBA data
cbaTab <- filter(fullTab, assay == "CBA") %>%
mutate(logVal = glog2(value)) %>%
mutate(Group = factor(Group, levels = c("no irAE","rheumatic irAE","other irAE")))Baseline samples
subTab <- filter(cbaTab, condition == "Baseline" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()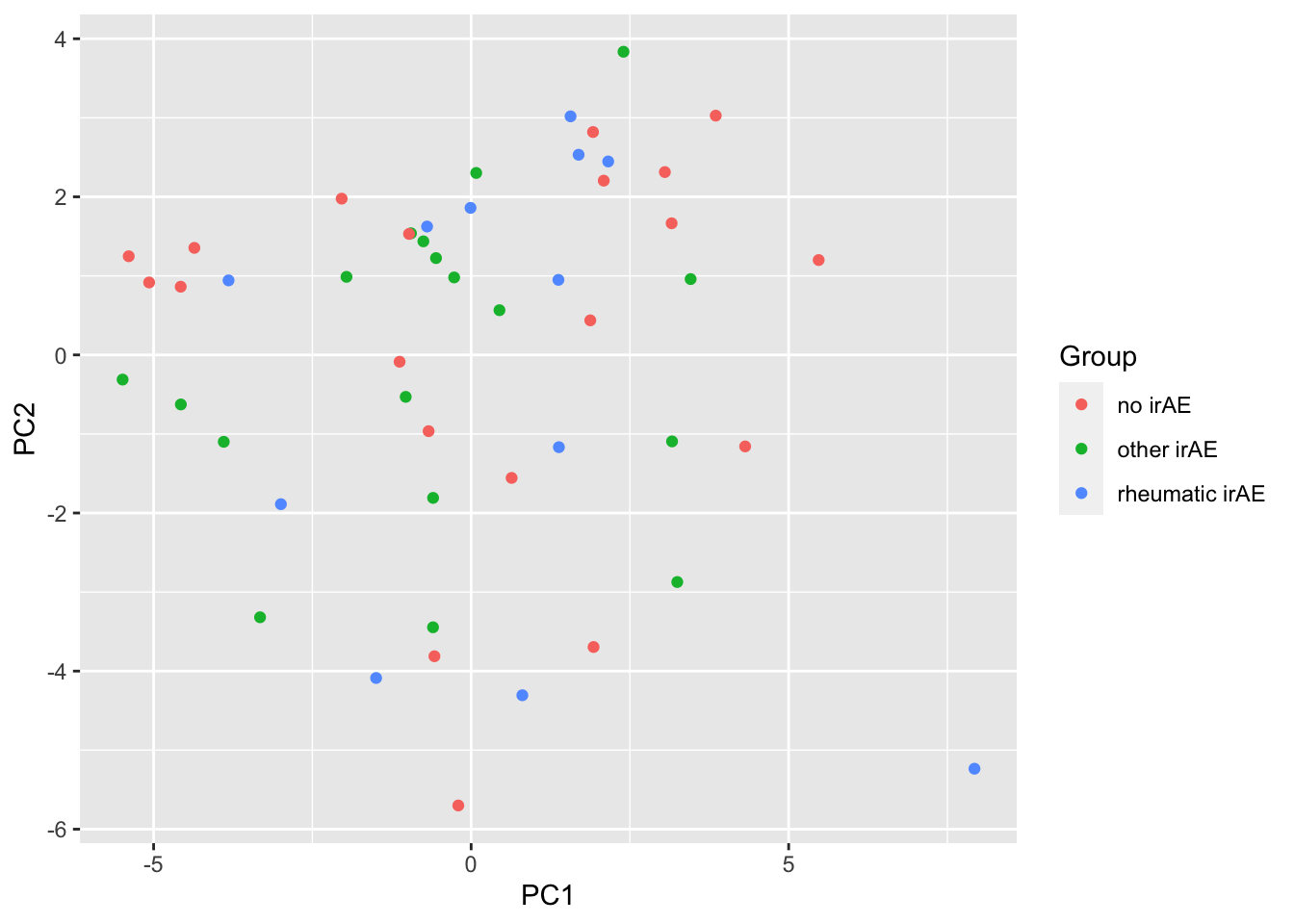
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC3 0.0162
2 PC4 0.0267PC3 versus PC4
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()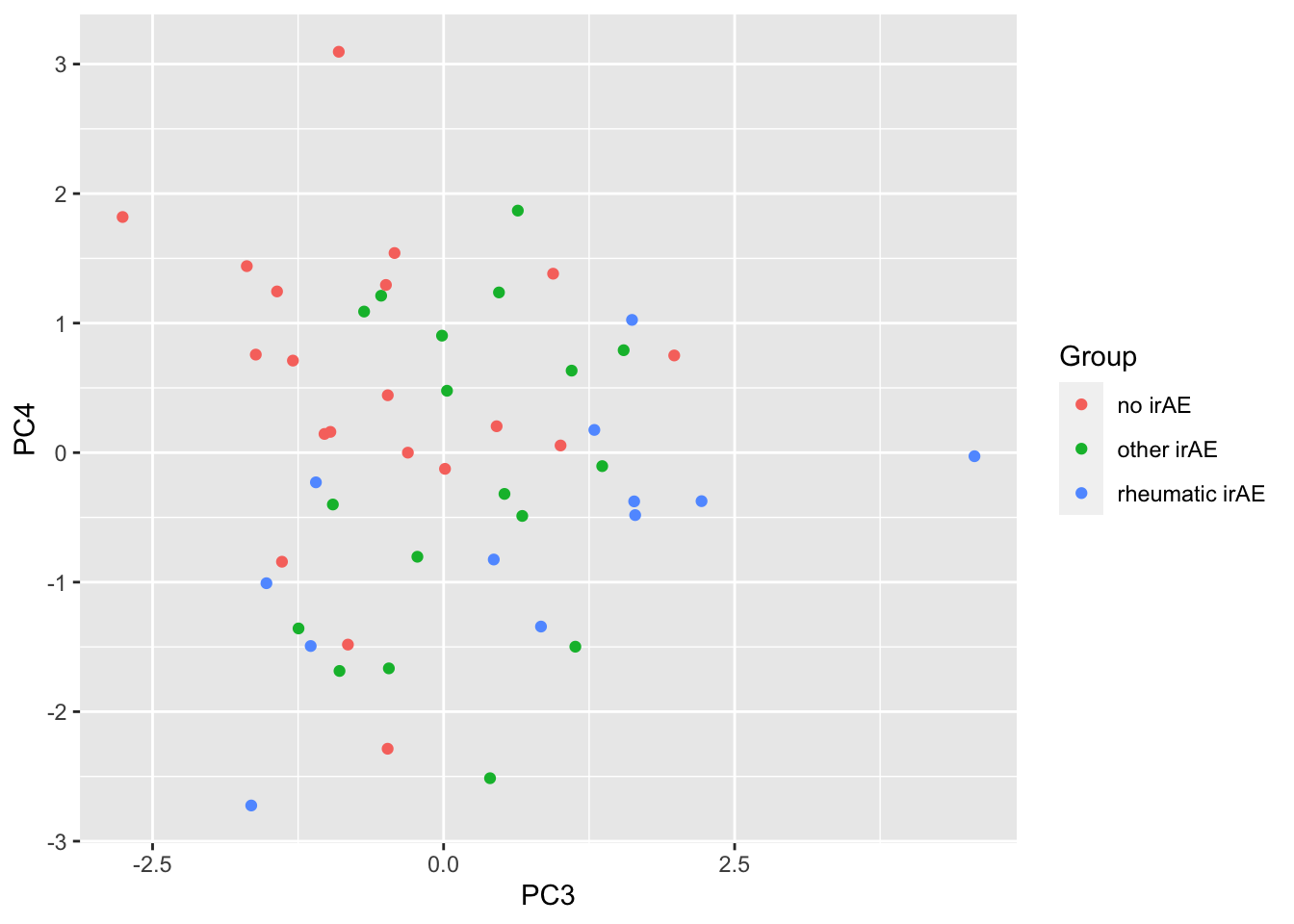
ANOVA test
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 MCP-1 0.0163Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")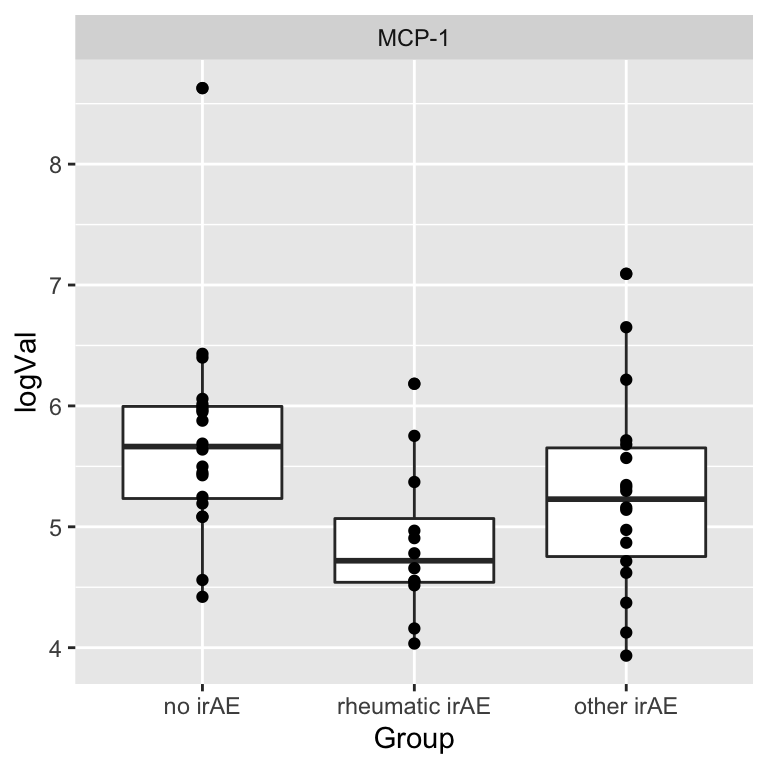
Pair-wise comparison between phenotypes
pairwiseT <- function(dataTab, value, group, feature) {
if (is.factor(dataTab[[group]])) {
allCompare <- combn(levels(dataTab[[group]]),2, simplify = FALSE)
} else {
allCompare <- combn(unique(dataTab[[group]]),2, simplify = FALSE)
}
testTab <- select(dataTab, all_of(c(value, group, feature))) %>%
dplyr::rename(group = all_of(group),
value = all_of(value),
feature = all_of(feature))
resTab <- lapply(allCompare, function(x) {
refGroup <- x[1]
comGroup <- x[2]
eachTab <- filter(testTab, group %in% c(refGroup, comGroup)) %>%
mutate(group = factor(group, levels = c(refGroup, comGroup)))
res <- group_by(eachTab, feature) %>% nest() %>%
mutate(m = map(data, ~t.test(value~group,.,var.equal=TRUE))) %>%
mutate(r = map(m, broom::tidy)) %>% unnest(r) %>%
select(feature, estimate, p.value) %>%
mutate(group1 = refGroup, group2 = comGroup) %>%
arrange(p.value) %>% ungroup() %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))
}) %>% bind_rows()
}
resPair.baseline <- pairwiseT(subTab, "logVal", "Group", "name")
filter(resPair.baseline, p.value < 0.05)# A tibble: 4 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 MCP-1 0.861 0.00563 no irAE rheumatic irAE 0.146
2 IL-18 0.824 0.0381 no irAE other irAE 0.514
3 IL-17A 0.526 0.0444 no irAE other irAE 0.514
4 TNF-alpha 1.22 0.0496 rheumatic irAE other irAE 0.701Follow-up samples
subTab <- filter(cbaTab, condition == "Follow_Up" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()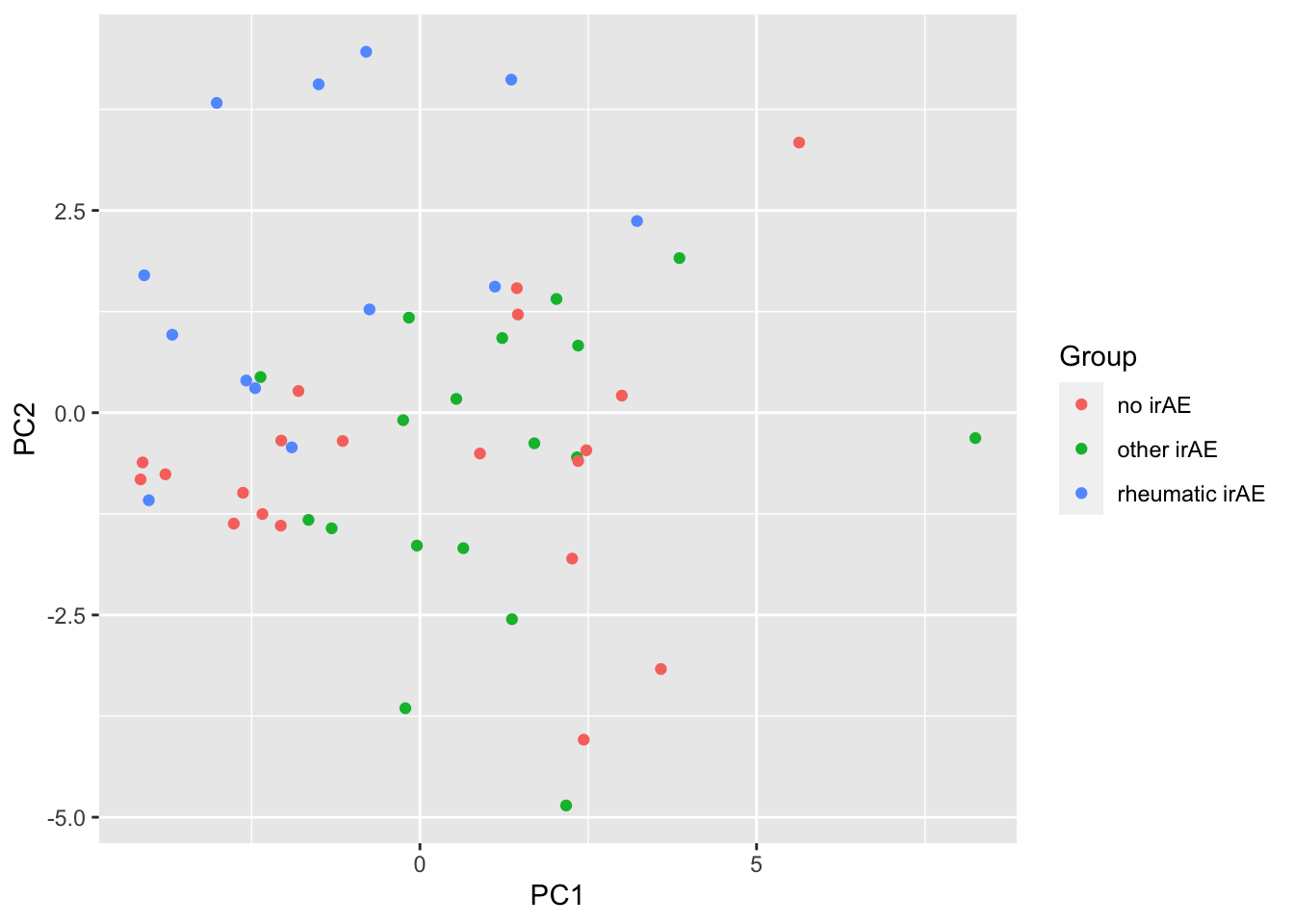
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()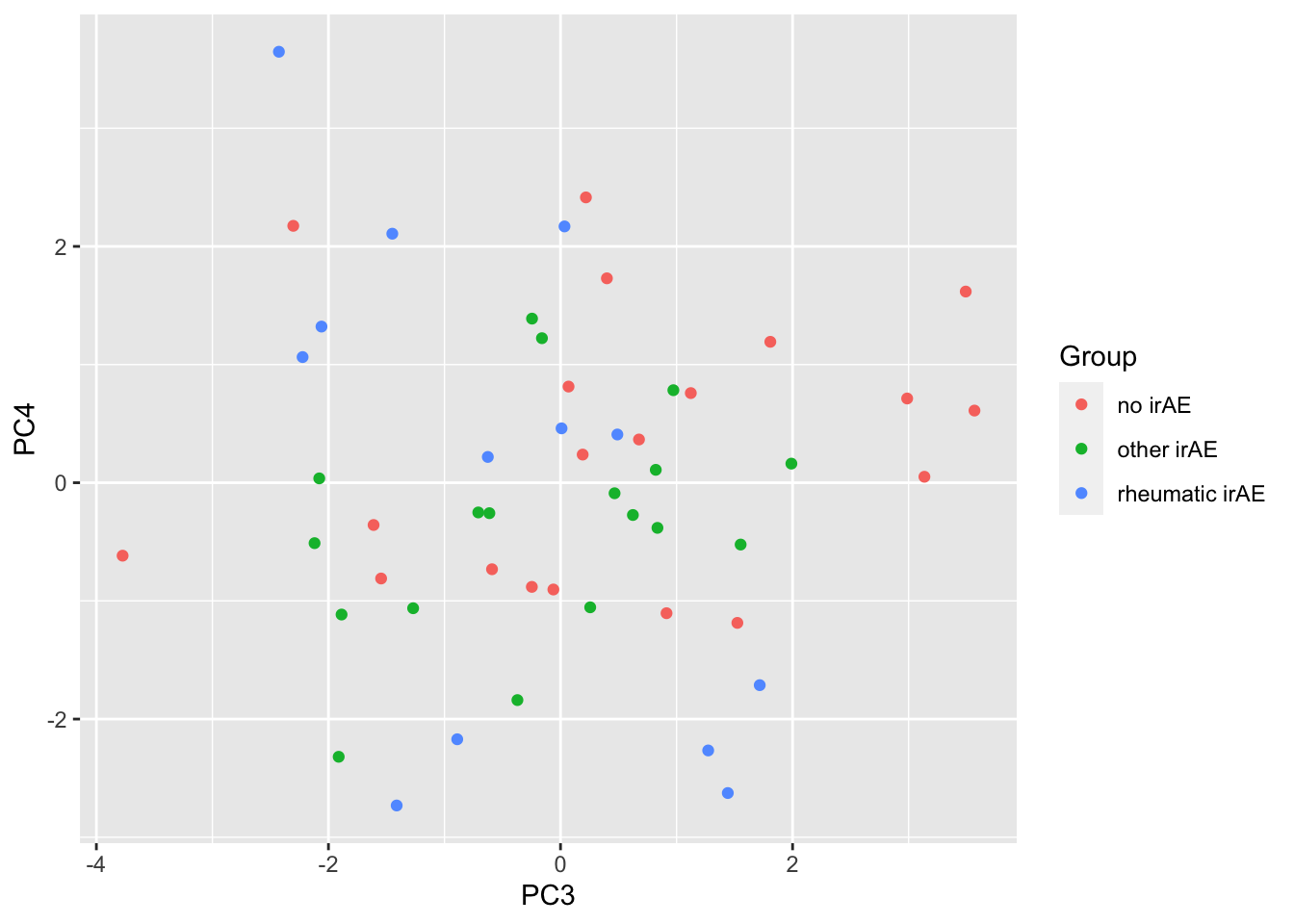
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC2 0.000295
2 PC1 0.0280 ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 11 × 2
# Groups: name [11]
name p.value
<chr> <dbl>
1 IL-33 0.0000484
2 IL-10 0.000209
3 IL-12p70 0.000258
4 IL-1beta 0.00821
5 Perforin 0.0113
6 IFN-alpha2 0.0138
7 IFN-gamma 0.0148
8 IL-6 0.0276
9 IL-17A 0.0296
10 IL-23 0.0304
11 GM-CSF 0.0388 Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")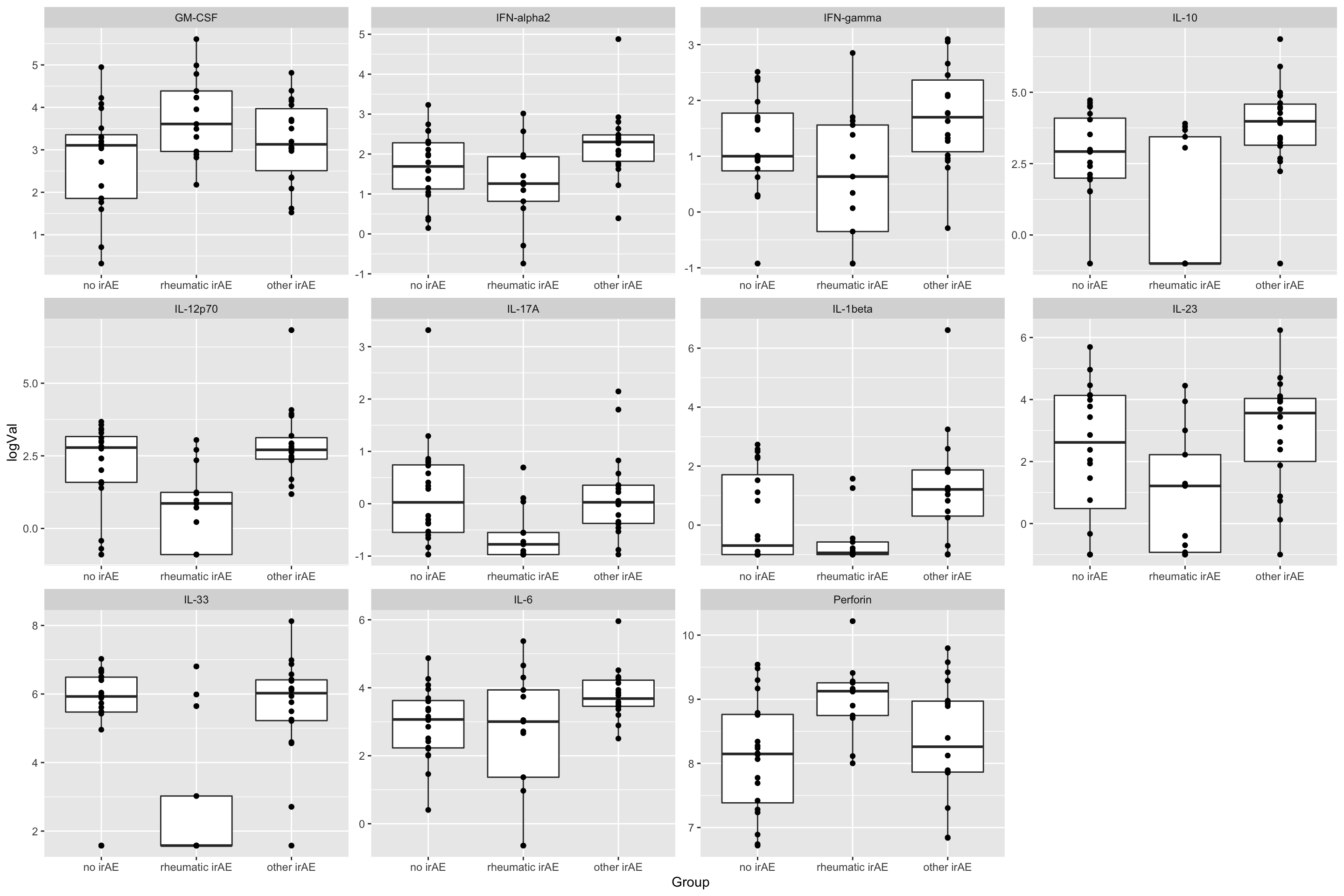
Pair-wise comparison
resPair.followup <- pairwiseT(subTab, "logVal", "Group", "name")
filter(resPair.followup, p.value < 0.05)# A tibble: 19 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 IL-33 2.67 0.000294 no irAE rheumatic irAE 0.00764
2 Perforin -0.915 0.00242 no irAE rheumatic irAE 0.0315
3 IL-12p70 1.48 0.00612 no irAE rheumatic irAE 0.0530
4 IL-10 1.94 0.00830 no irAE rheumatic irAE 0.0540
5 GM-CSF -0.992 0.0175 no irAE rheumatic irAE 0.0822
6 IL-17A 0.755 0.0190 no irAE rheumatic irAE 0.0822
7 IL-6 -0.878 0.00581 no irAE other irAE 0.151
8 Granzyme -2.86 0.0476 no irAE other irAE 0.345
9 IL-12p70 -2.17 0.0000880 rheumatic irAE other irAE 0.00125
10 IL-33 -2.86 0.0000964 rheumatic irAE other irAE 0.00125
11 IL-10 -3.02 0.000224 rheumatic irAE other irAE 0.00194
12 IL-1beta -1.77 0.00316 rheumatic irAE other irAE 0.0205
13 IL-23 -2.01 0.00665 rheumatic irAE other irAE 0.0272
14 IFN-gamma -1.07 0.00728 rheumatic irAE other irAE 0.0272
15 IFN-alpha2 -0.997 0.00754 rheumatic irAE other irAE 0.0272
16 IL-17A -0.733 0.00837 rheumatic irAE other irAE 0.0272
17 IL-6 -1.15 0.0259 rheumatic irAE other irAE 0.0749
18 Perforin 0.642 0.0315 rheumatic irAE other irAE 0.0819
19 IL-4 2.25 0.0475 rheumatic irAE other irAE 0.112 Use difference between follow_up and baseline
In this analysis, instead of looking at baseline and follow up separately, I created new features by using: follow_up - baseline, which indicates the change over time.
subTab <- filter(cbaTab, assay == "CBA", condition %in% c("Baseline", "Follow_Up")) %>%
select(name, Group, condition, logVal, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
mutate(diffVal = Follow_Up - Baseline) %>%
filter(!is.na(diffVal))PCA
subMat <- subTab %>% select(patID, name, diffVal) %>%
pivot_wider(names_from = name, values_from = diffVal) %>%
column_to_rownames("patID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "patID") %>%
left_join(patAnno, by = "patID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()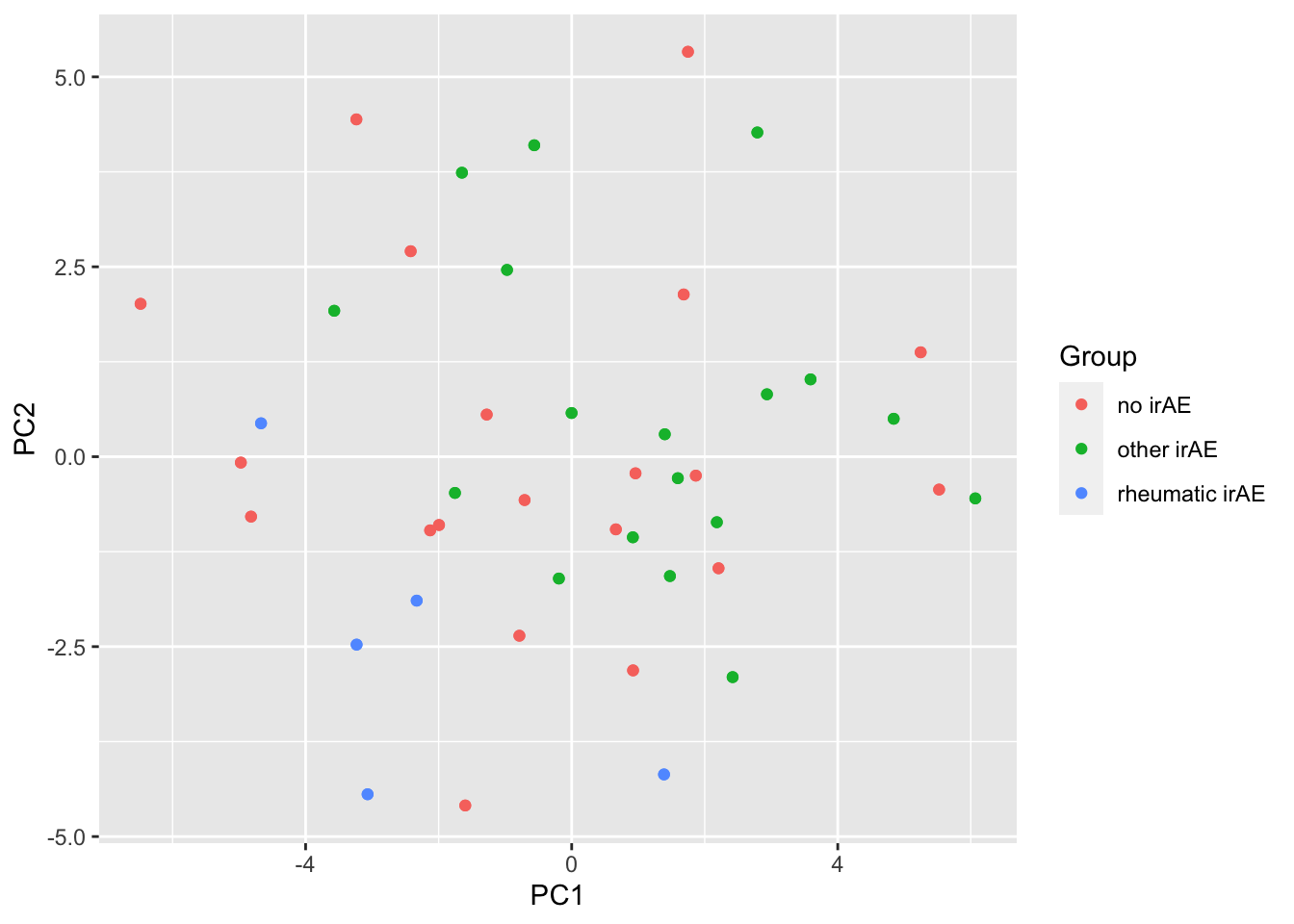
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()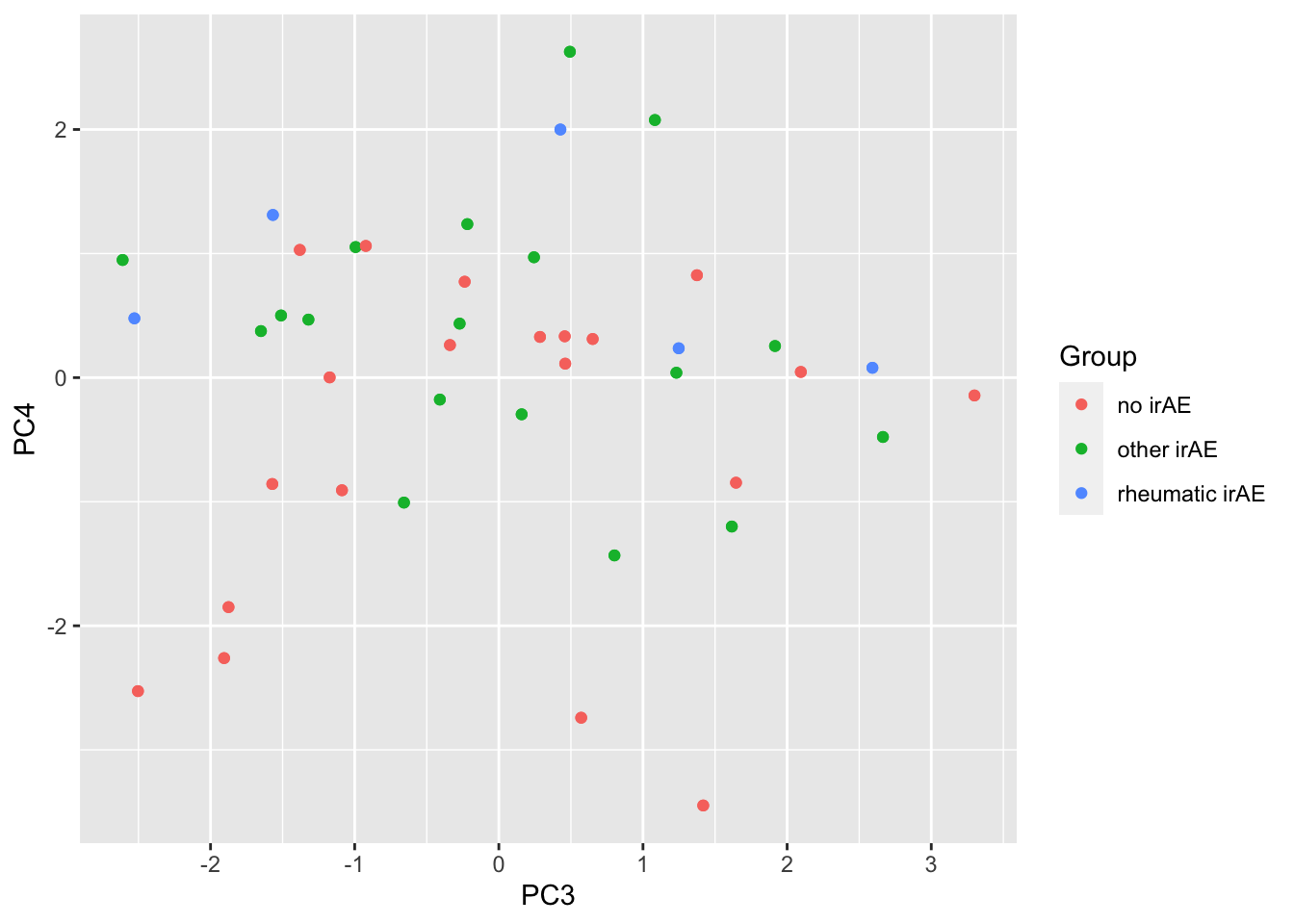
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 6 × 2
# Groups: name [6]
name p.value
<chr> <dbl>
1 PC4 0.000585
2 PC2 0.000609
3 PC1 0.000788
4 PC8 0.0145
5 PC12 0.0194
6 PC5 0.0426 PC4 versus PC2
ggplot(pcTab, aes(x=PC2, y=PC4, col = Group)) +
geom_point()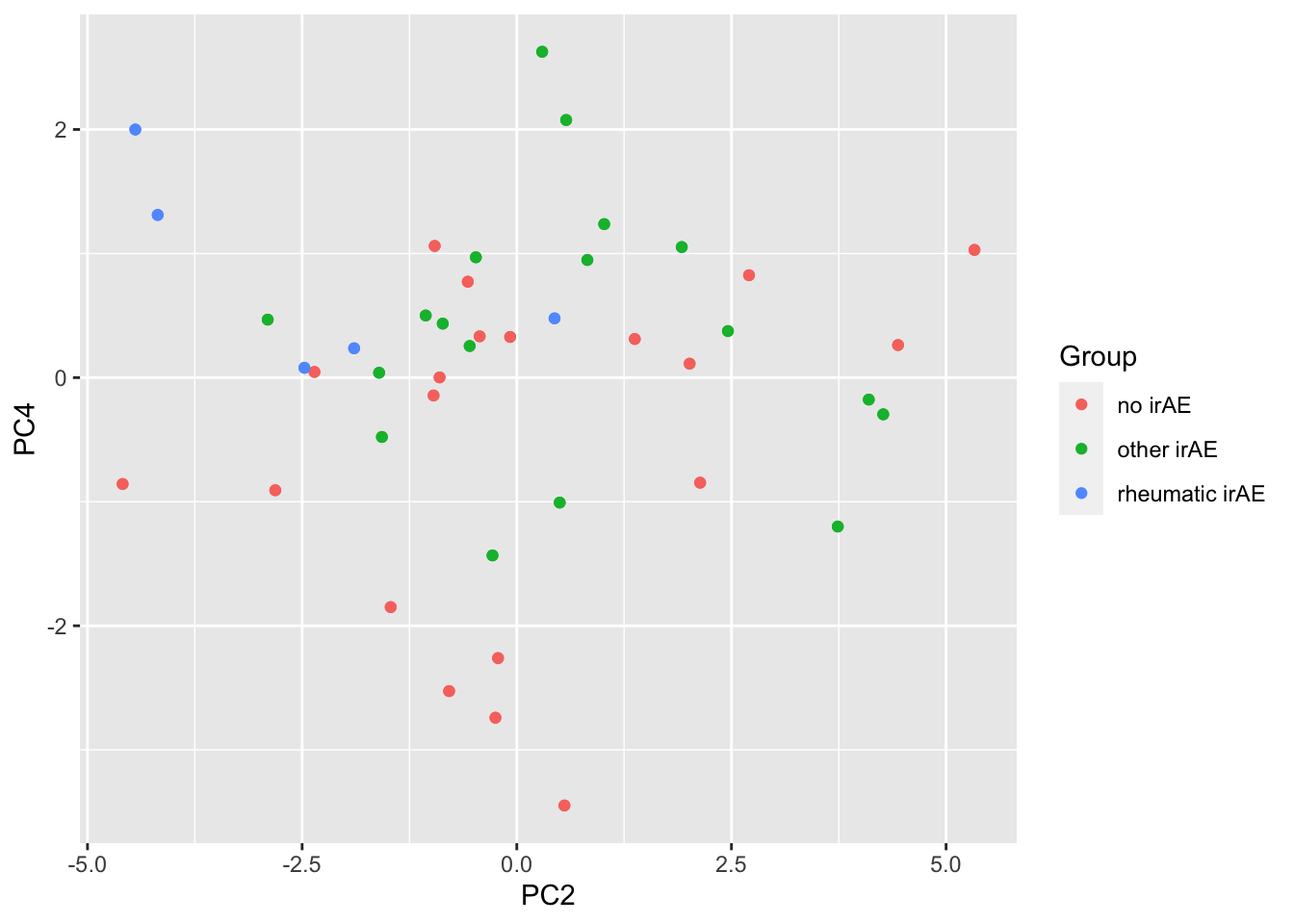
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(diffVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value <= 0.05)
resTab.sig# A tibble: 11 × 2
# Groups: name [11]
name p.value
<chr> <dbl>
1 IL-12p70 0.00215
2 IL-33 0.00258
3 IL-23 0.00527
4 IL-1beta 0.00852
5 IL-18 0.00857
6 IFN-alpha2 0.0118
7 IL-17A 0.0159
8 IL-4 0.0205
9 IL-10 0.0265
10 IFN-gamma 0.0393
11 TNF-alpha 0.0480 Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=diffVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")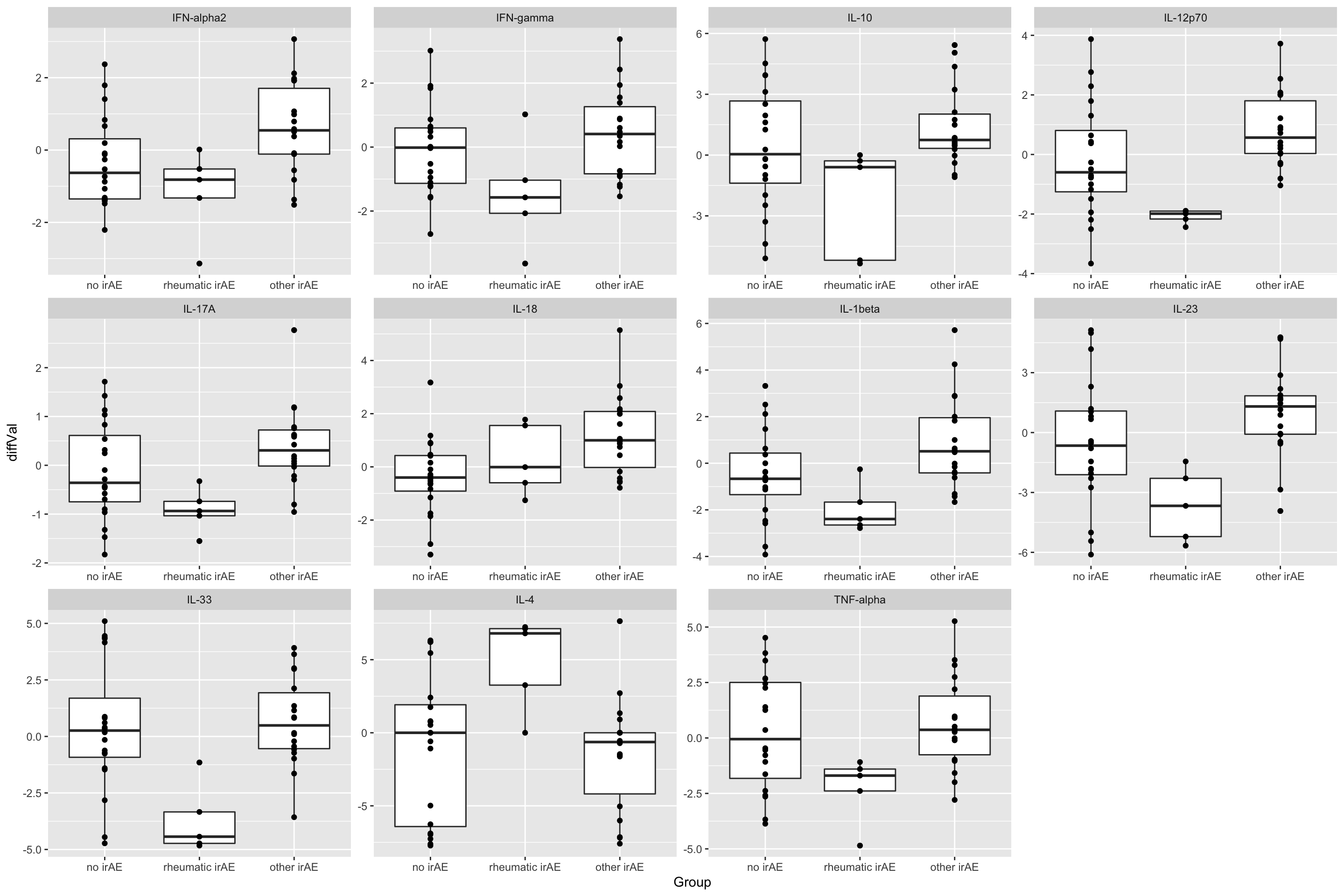
Pair-wise comparison
resPair.diff <- pairwiseT(subTab, "diffVal", "Group", "name")
filter(resPair.diff, p.value <= 0.05)# A tibble: 19 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 IL-33 4.12 0.00529 no irAE rheumatic irAE 0.138
2 IL-4 -5.83 0.0213 no irAE rheumatic irAE 0.250
3 IL-12p70 1.90 0.0371 no irAE rheumatic irAE 0.250
4 IL-23 3.15 0.0458 no irAE rheumatic irAE 0.250
5 TNF-alpha 2.55 0.0480 no irAE rheumatic irAE 0.250
6 IL-18 -1.56 0.00254 no irAE other irAE 0.0660
7 IFN-alpha2 -0.943 0.0260 no irAE other irAE 0.289
8 IL-1beta -1.41 0.0334 no irAE other irAE 0.289
9 IL-12p70 -2.89 0.0000528 rheumatic irAE other irAE 0.00137
10 IL-33 -4.36 0.000165 rheumatic irAE other irAE 0.00215
11 IL-23 -4.62 0.000318 rheumatic irAE other irAE 0.00275
12 IL-10 -3.68 0.00247 rheumatic irAE other irAE 0.0142
13 IL-17A -1.31 0.00320 rheumatic irAE other irAE 0.0142
14 IL-4 6.34 0.00327 rheumatic irAE other irAE 0.0142
15 IL-1beta -2.85 0.00663 rheumatic irAE other irAE 0.0246
16 TNF-alpha -2.95 0.00804 rheumatic irAE other irAE 0.0261
17 IFN-alpha2 -1.75 0.0118 rheumatic irAE other irAE 0.0341
18 IFN-gamma -1.88 0.0172 rheumatic irAE other irAE 0.0447
19 IL-8 -3.12 0.0395 rheumatic irAE other irAE 0.0934 Summarise the pariwise comparison results using a heatmap
resPair.cba <- bind_rows(
resPair.baseline %>% mutate(condition = "Baseline"),
resPair.followup %>% mutate(condition = "Follow_Up"),
resPair.diff %>% mutate(condition = "Follow_Up - Baseline")
)
plotTab <- mutate(resPair.cba,
comparison = paste0(group2, " ~ ", group1),
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=feature, x=comparison)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
facet_wrap(~condition, ncol=3) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low = "blue", high="red", midpoint = 0)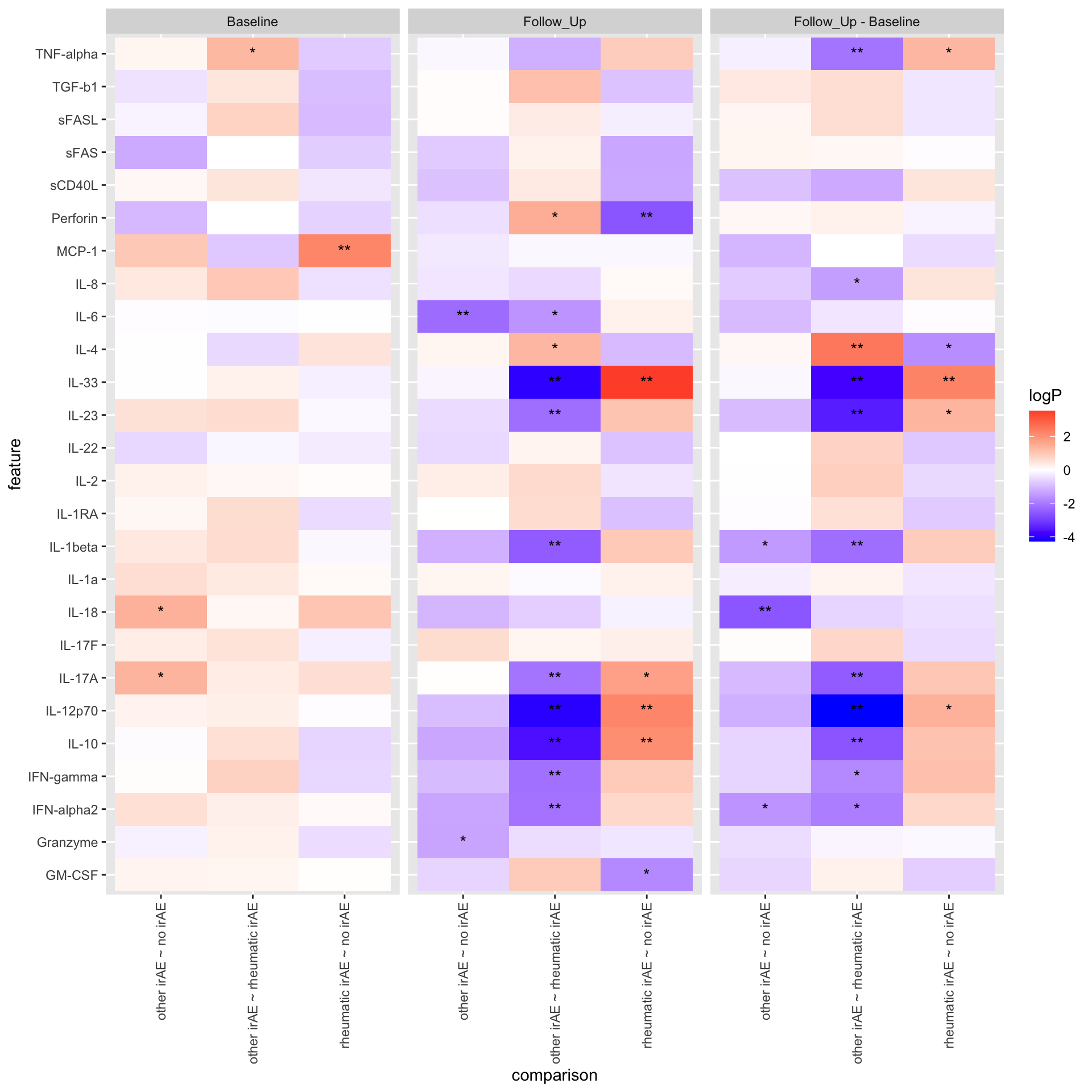
NMR data
nmrTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = value) %>%
mutate(Group = factor(Group, levels = c("no irAE","rheumatic irAE","other irAE")))Try vsn transformation (not used, may introduce artefact)
nmrMat <- assays(mae)[["nmr"]]
#vsnFit <- vsn::vsnMatrix(nmrMat, minDataPointsPerStratum = 10)
#nmrMat <- predict(vsnFit, nmrMat)
vsnTab <- nmrMat %>% as_tibble(rownames = "name") %>%
pivot_longer(-name, names_to = "sampleID", values_to = "logVal")
#nmrTab <- left_join(nmrTab, vsnTab, by =c("sampleID","name"))Baseline samples
subTab <- filter(nmrTab, condition == "Baseline" )PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()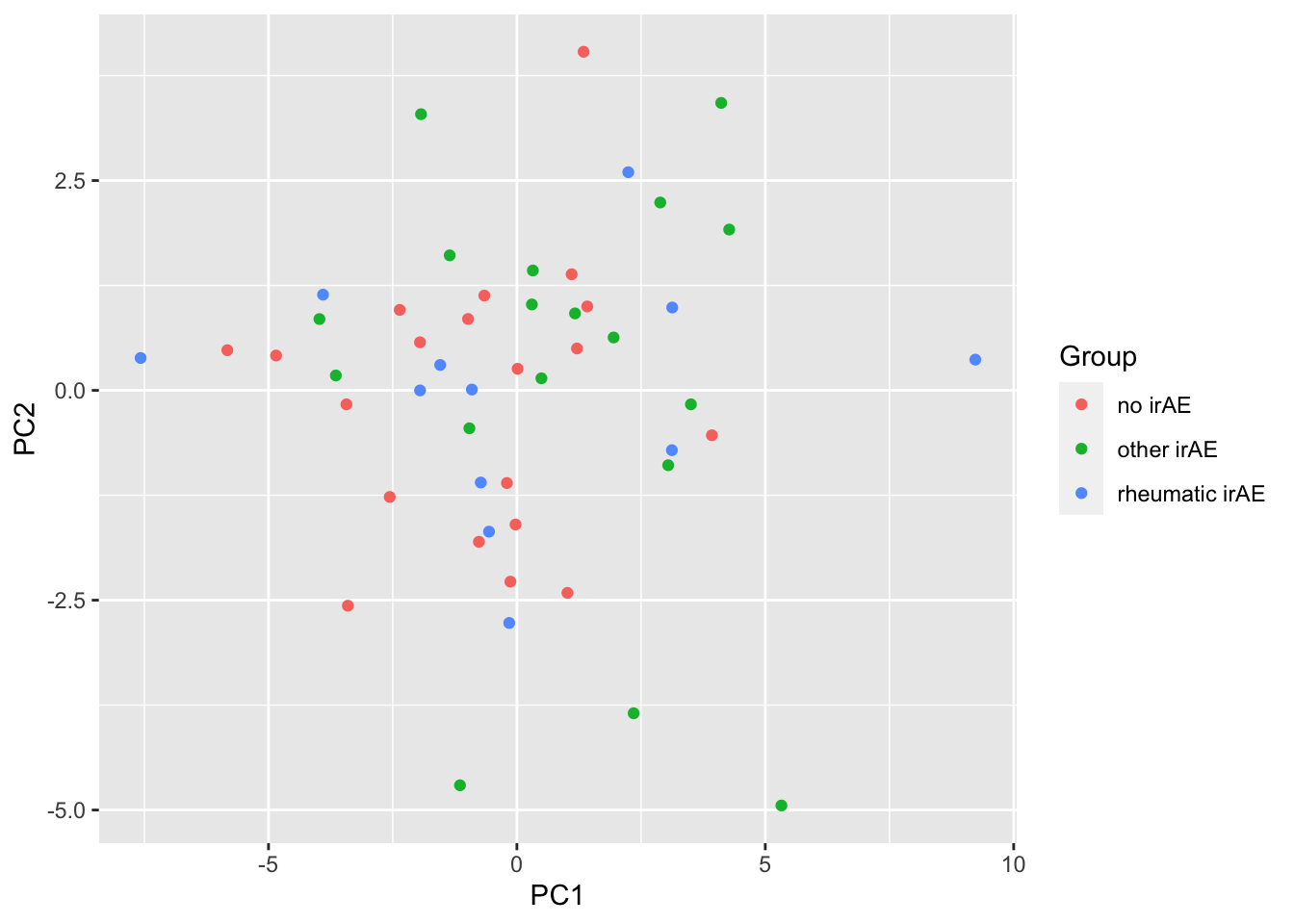
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()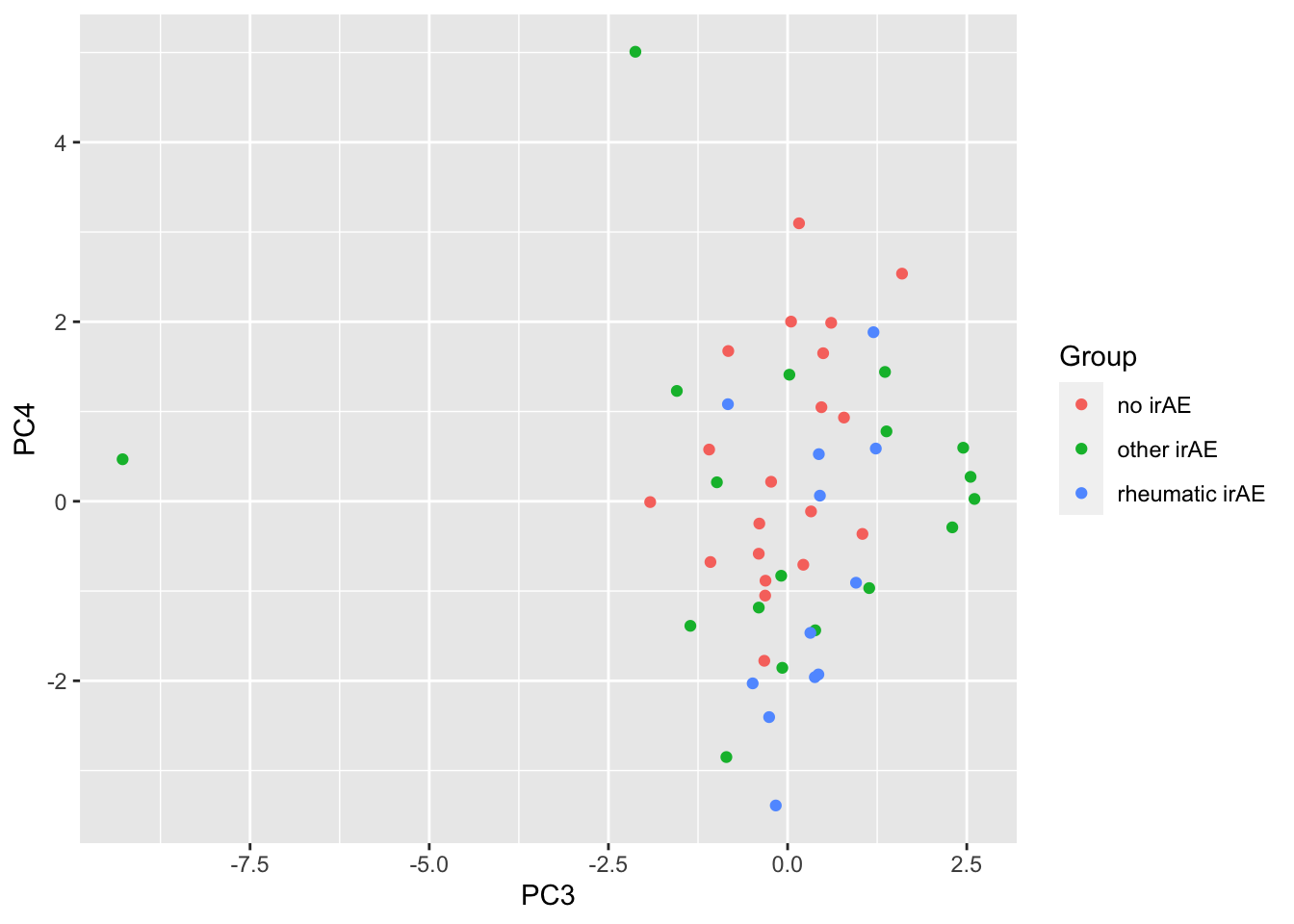
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC12 0.0132
2 PC11 0.0162ggplot(pcTab, aes(x=PC11, y=PC12, col = Group)) +
geom_point()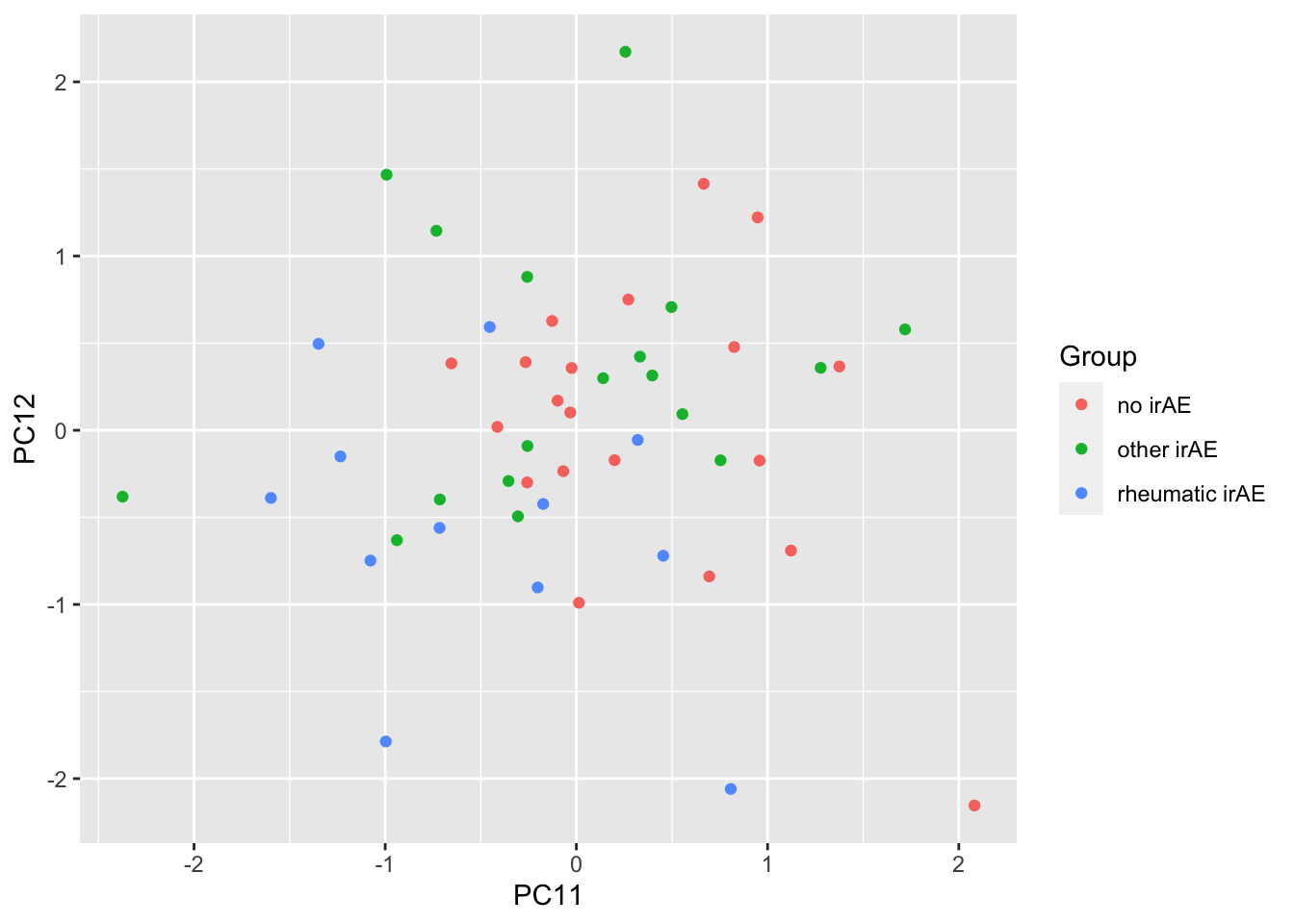
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 Histidine 0.000554Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")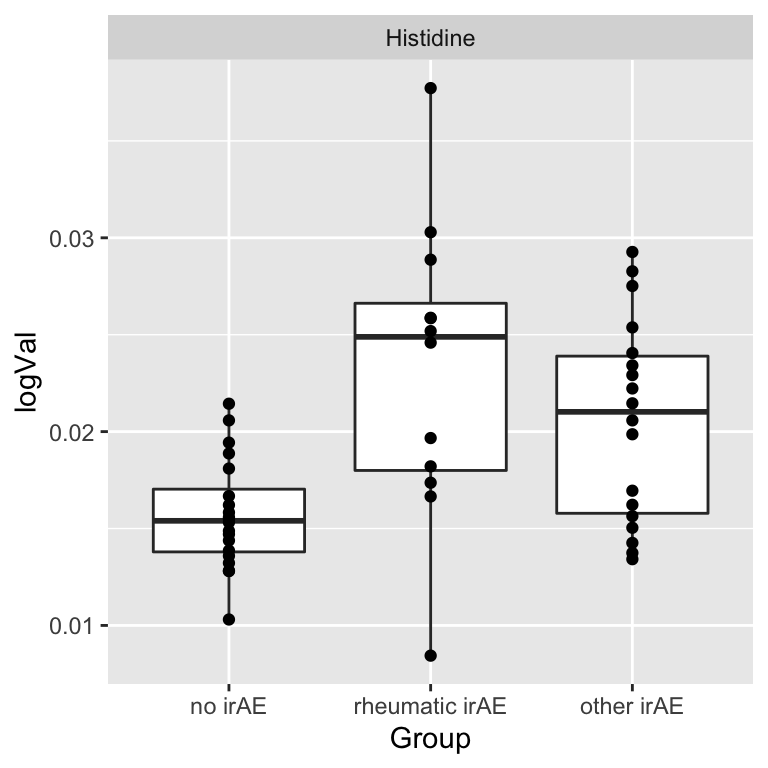
Pair-wise comparison
resPair.baseline <- pairwiseT(subTab, "logVal", "Group", "name")
filter(resPair.baseline, p.value < 0.05)# A tibble: 6 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 Histidine -0.00752 0.000384 no irAE rheumatic irAE 0.0127
2 Alanine -0.0248 0.0379 no irAE rheumatic irAE 0.603
3 Histidine -0.00486 0.000869 no irAE other irAE 0.0287
4 Asparagine -0.00672 0.0219 no irAE other irAE 0.362
5 Leucine -0.0107 0.0349 no irAE other irAE 0.384
6 Succinate -0.00150 0.0357 rheumatic irAE other irAE 0.857 Follow-up samples
subTab <- filter(nmrTab, condition == "Follow_Up" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()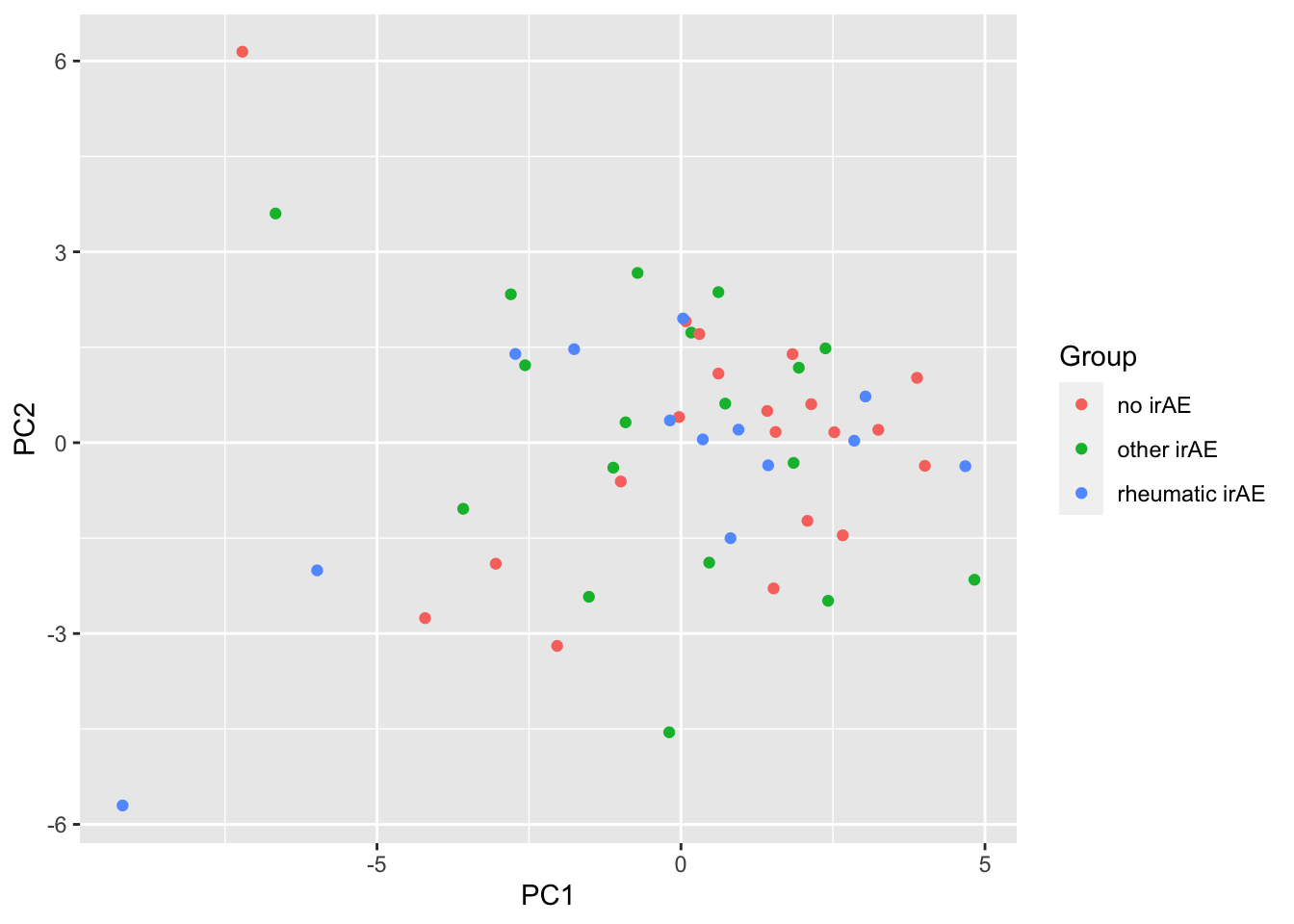
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()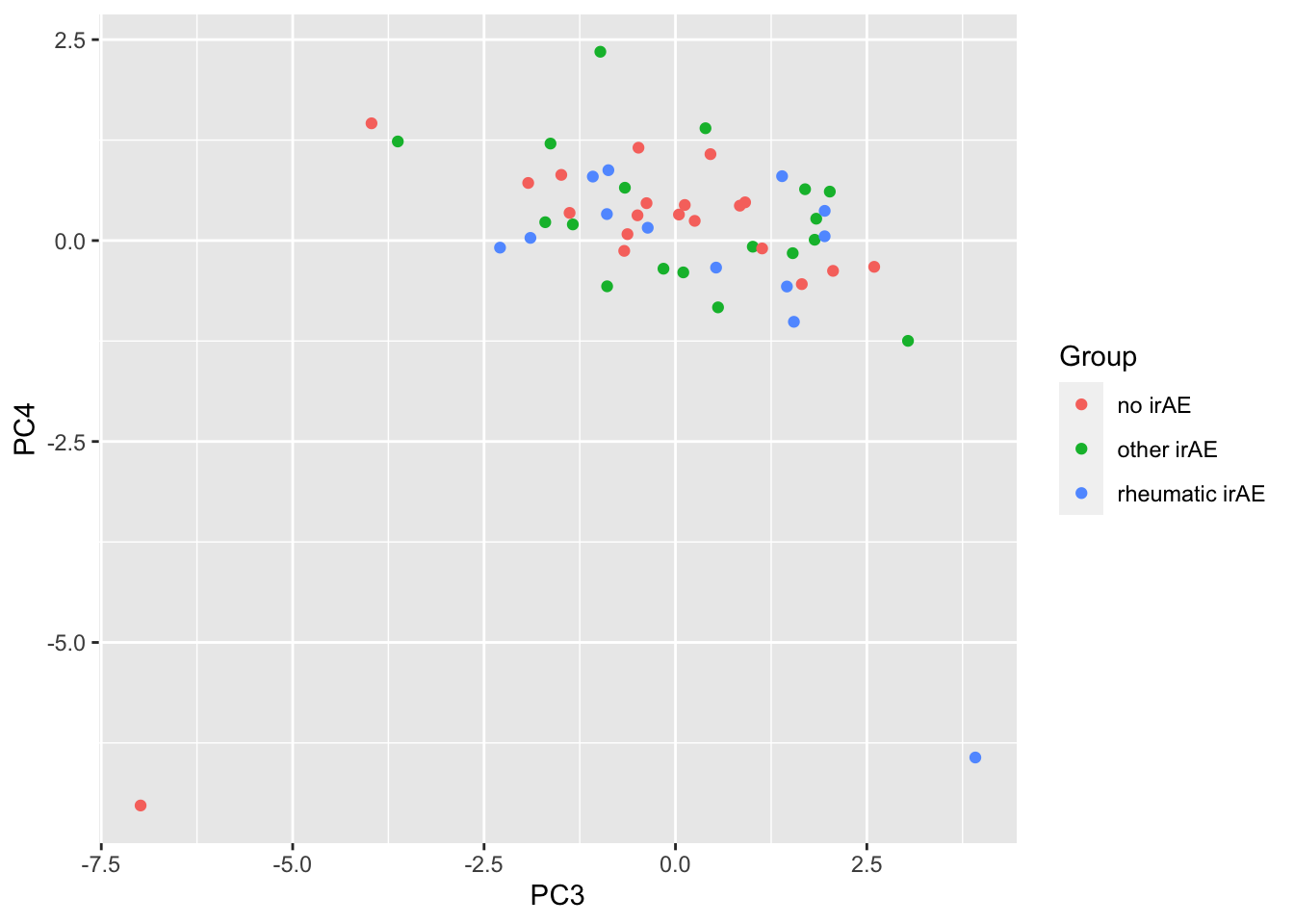
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC11 0.00789
2 PC13 0.0341 ggplot(pcTab, aes(x=PC11, y=PC13, col = Group)) +
geom_point()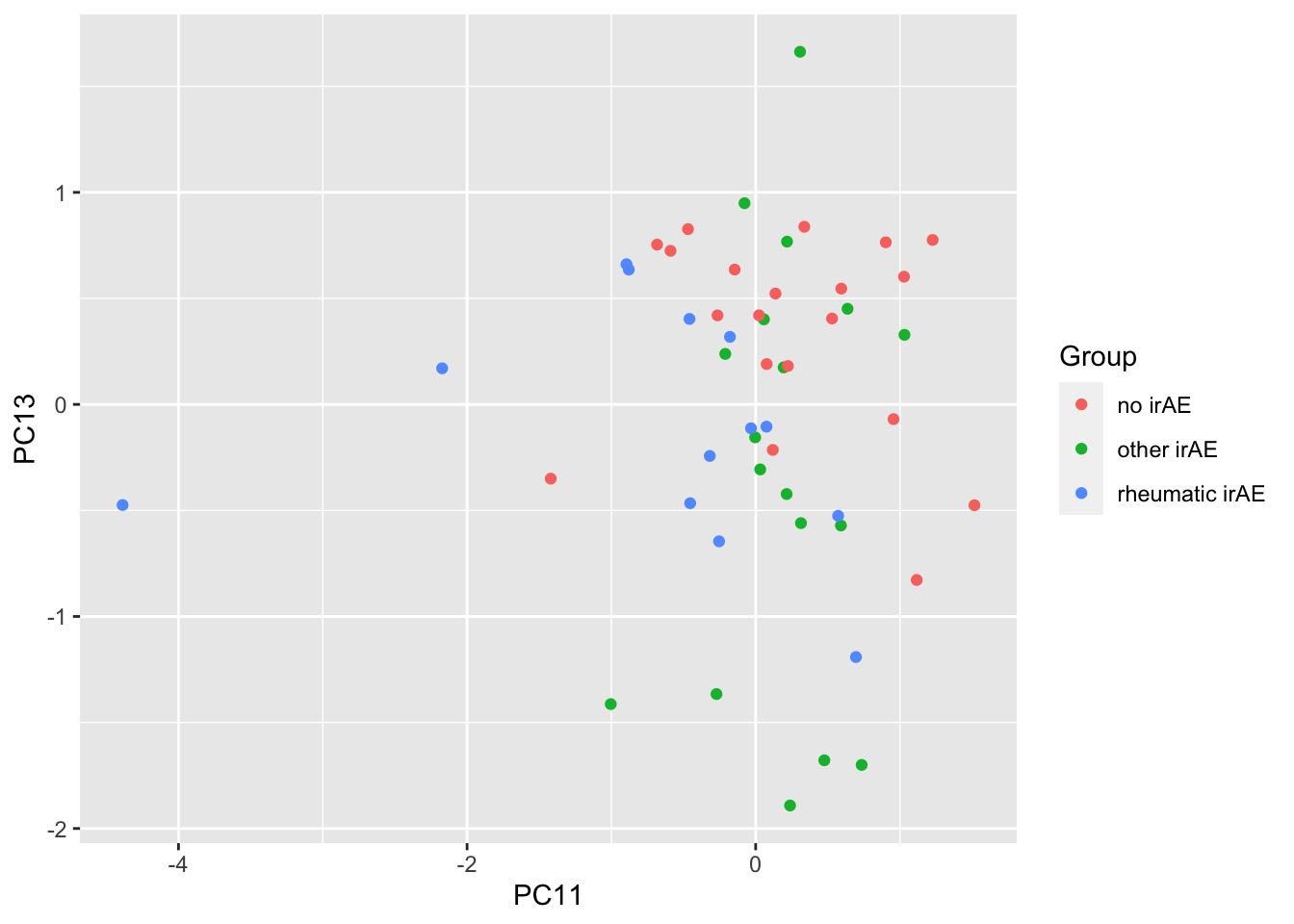
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 Histidine 0.0385
2 Glutamine 0.0454Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")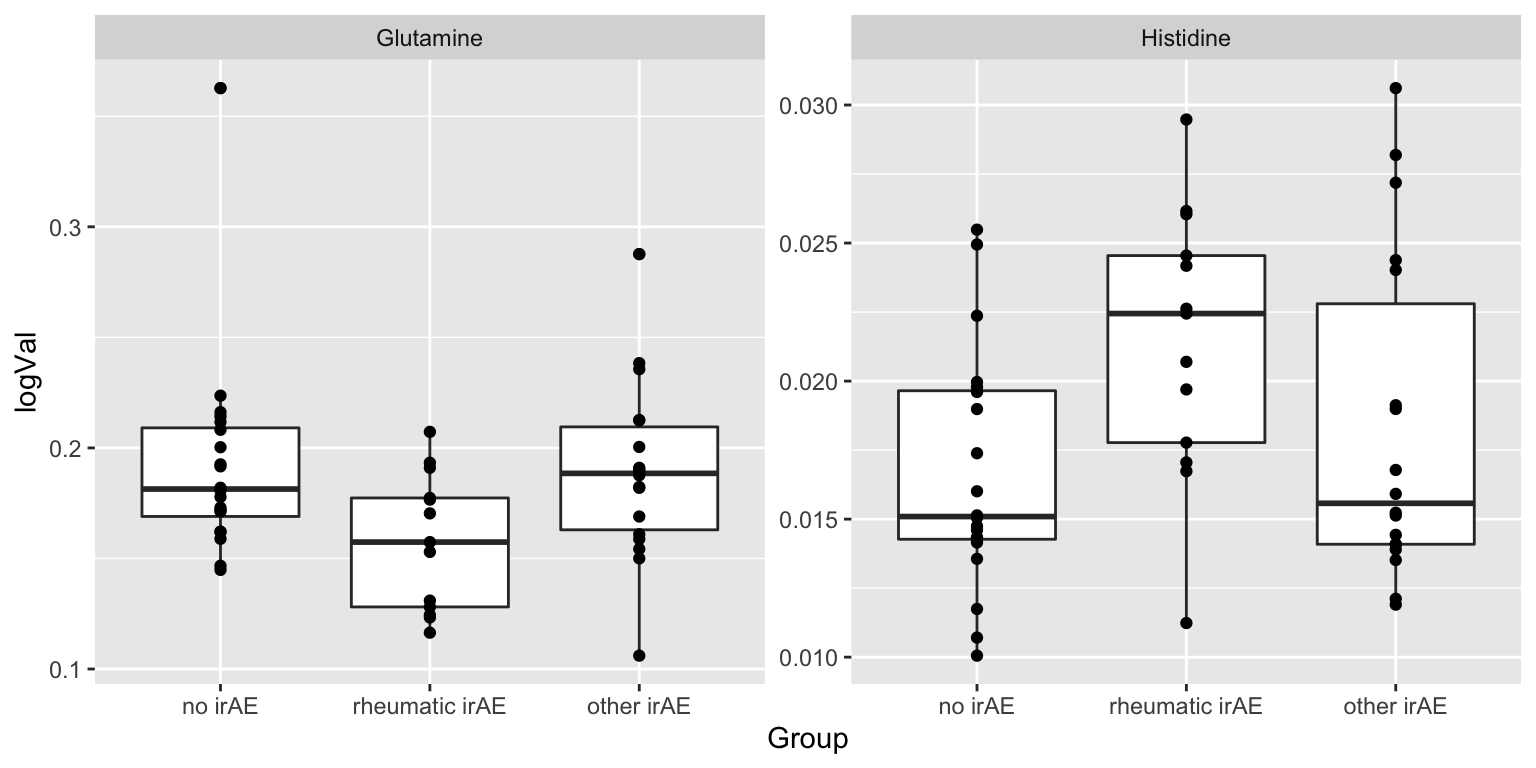
Pair-wise comparison
resPair.followup <- pairwiseT(subTab, "logVal", "Group", "name")
filter(resPair.followup, p.value < 0.05)# A tibble: 4 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 Histidine -0.00479 0.00615 no irAE rheumatic irAE 0.203
2 Glutamine 0.0350 0.0228 no irAE rheumatic irAE 0.377
3 Acetate -0.00509 0.0409 no irAE rheumatic irAE 0.450
4 Glutamine -0.0317 0.0238 rheumatic irAE other irAE 0.786Use difference between follow_up and baseline
subTab <- filter(nmrTab, condition %in% c("Baseline", "Follow_Up")) %>%
select(name, Group, condition, logVal, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
mutate(diffVal = Follow_Up - Baseline) %>%
filter(!is.na(diffVal))PCA
subMat <- subTab %>% select(patID, name, diffVal) %>%
pivot_wider(names_from = name, values_from = diffVal) %>%
column_to_rownames("patID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "patID") %>%
left_join(patAnno, by = "patID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()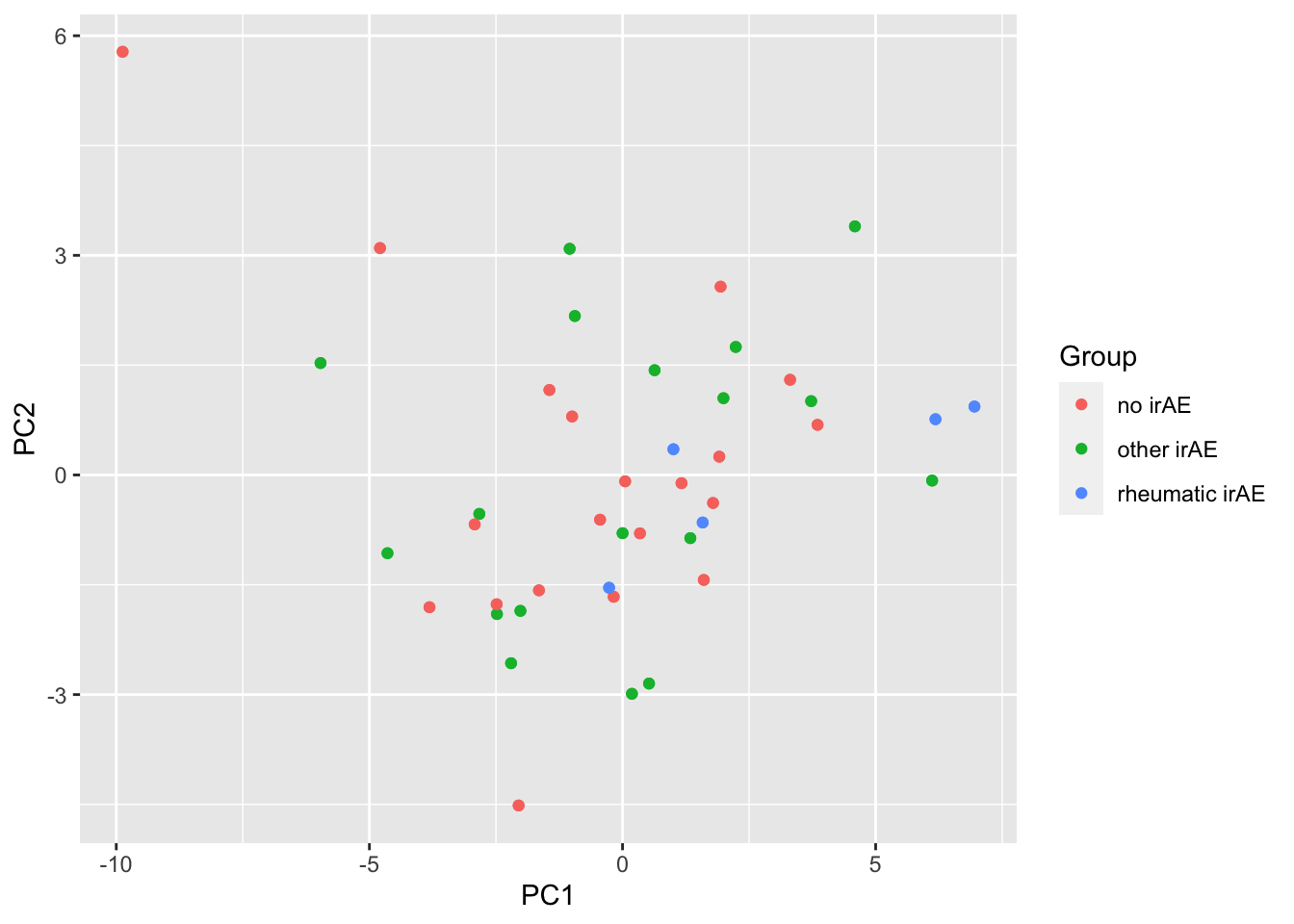
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 9 × 2
# Groups: name [9]
name p.value
<chr> <dbl>
1 PC14 0.00146
2 PC1 0.00335
3 PC4 0.0131
4 PC3 0.0134
5 PC16 0.0217
6 PC12 0.0317
7 PC20 0.0352
8 PC7 0.0356
9 PC6 0.0446 ggplot(pcTab, aes(x=PC1, y=PC14, col = Group)) +
geom_point()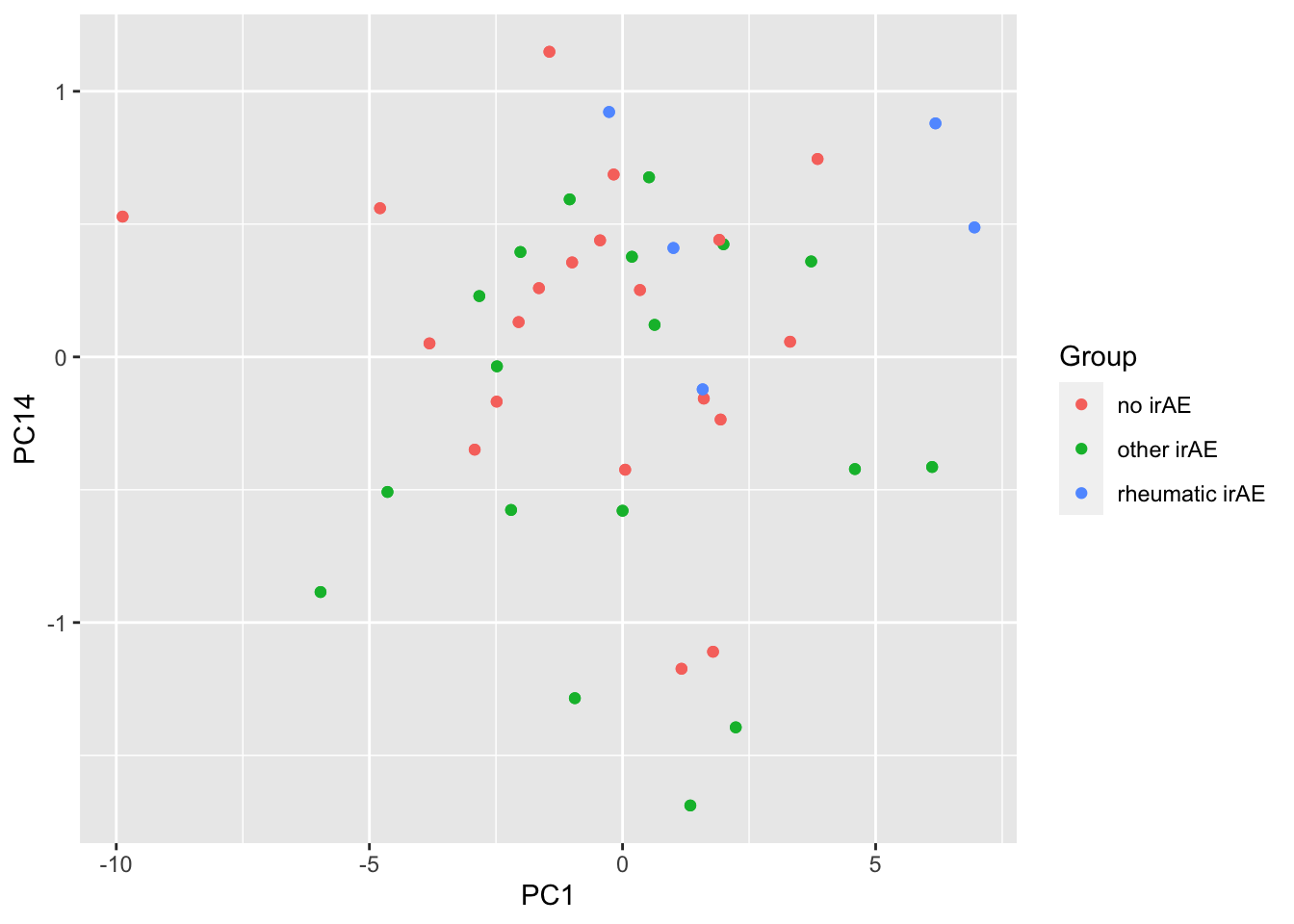
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(diffVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 6 × 2
# Groups: name [6]
name p.value
<chr> <dbl>
1 Choline 0.0137
2 Alanine 0.0140
3 Leucine 0.0182
4 Valine 0.0238
5 Histidine 0.0277
6 Asparagine 0.0380Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=diffVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")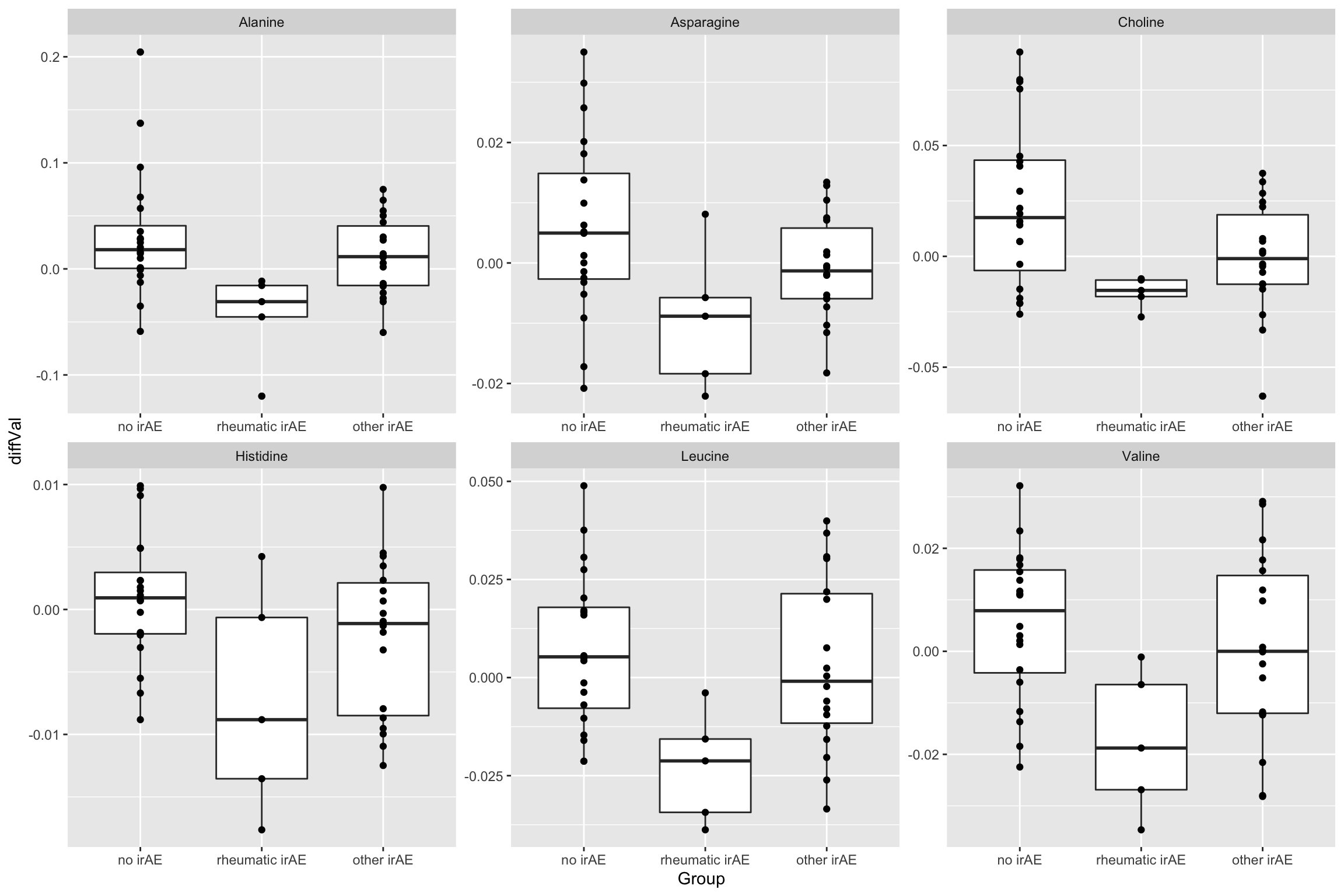
Pair-wise comparison
resPair.diff <- pairwiseT(subTab, "diffVal", "Group", "name")
filter(resPair.diff, p.value < 0.05)# A tibble: 14 × 6
feature estimate p.value group1 group2 p.adj
<chr> <dbl> <dbl> <chr> <chr> <dbl>
1 Leucine 0.0303 0.00408 no irAE rheumatic irAE 0.0691
2 Valine 0.0229 0.00419 no irAE rheumatic irAE 0.0691
3 Histidine 0.00822 0.0114 no irAE rheumatic irAE 0.115
4 Alanine 0.0761 0.0139 no irAE rheumatic irAE 0.115
5 Choline 0.0395 0.0284 no irAE rheumatic irAE 0.188
6 Proline 0.00988 0.0411 no irAE rheumatic irAE 0.202
7 Asparagine 0.0152 0.0439 no irAE rheumatic irAE 0.202
8 Choline 0.0238 0.0280 no irAE other irAE 0.796
9 Alanine -0.0569 0.00789 rheumatic irAE other irAE 0.186
10 Glutamine -0.0330 0.0113 rheumatic irAE other irAE 0.186
11 Leucine -0.0259 0.0231 rheumatic irAE other irAE 0.231
12 Tyrosine -0.00631 0.0280 rheumatic irAE other irAE 0.231
13 Glutamate -0.0271 0.0447 rheumatic irAE other irAE 0.259
14 Valine -0.0183 0.0471 rheumatic irAE other irAE 0.259 Summarise the pariwise comparison results using a heatmap
resPair.cba <- bind_rows(
resPair.baseline %>% mutate(condition = "Baseline"),
resPair.followup %>% mutate(condition = "Follow_Up"),
resPair.diff %>% mutate(condition = "Follow_Up - Baseline")
)
plotTab <- mutate(resPair.cba,
comparison = paste0(group2, " ~ ", group1),
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=feature, x=comparison)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
facet_wrap(~condition, ncol=3) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low = "blue", high="red", midpoint = 0)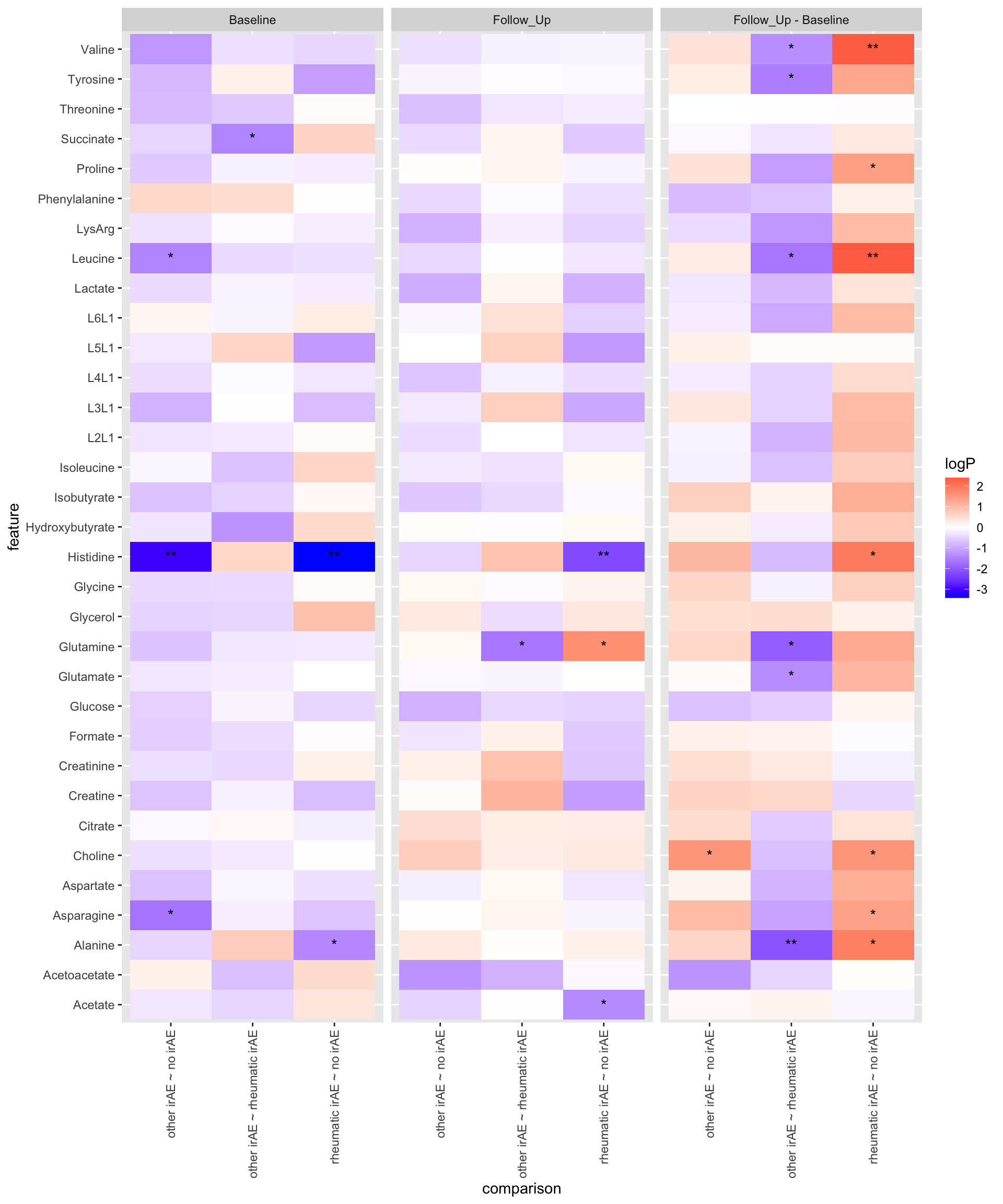
Focuse on cancer patients, combine rheumatic irAE and other irAE into one irAE group
CBA data
cbaTab <- filter(fullTab, assay == "CBA") %>%
mutate(logVal = glog2(value)) %>%
mutate(Group2 = ifelse(Group == "no irAE", Group, "irAE")) %>%
mutate(Group2 = factor(Group2, levels = c("no irAE","irAE")))
patAnno <- mutate(patAnno, Group2 = ifelse(Group == "no irAE", Group, "irAE"))Baseline samples
subTab <- filter(cbaTab, condition == "Baseline" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <= 0.05)# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 PC3 0.00923
2 PC4 0.0141
3 PC10 0.0423 ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 MCP-1 0.0104
2 IL-18 0.0153
3 IL-17A 0.0480Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=logVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")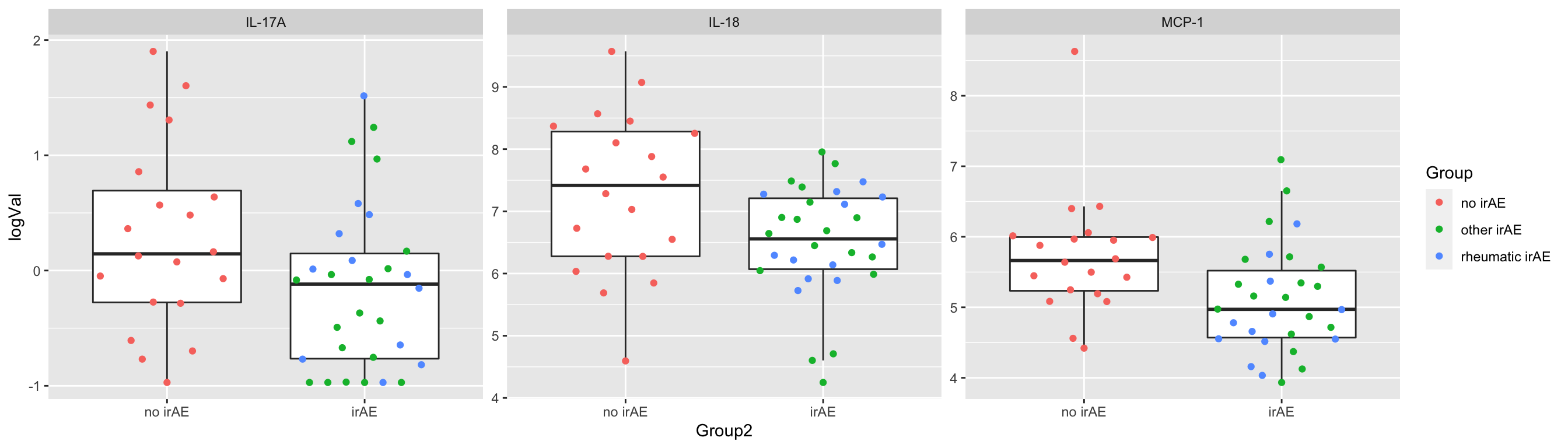
Pair-wise comparison
resPair.baseline <- pairwiseT(subTab, "logVal", "Group2", "name")
#filter(resPair.baseline, p.value < 0.05)Follow-up samples
subTab <- filter(cbaTab, condition == "Follow_Up" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 PC15 0.0274ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 Perforin 0.0313
2 sCD40L 0.0343
3 GM-CSF 0.0376Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=logVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")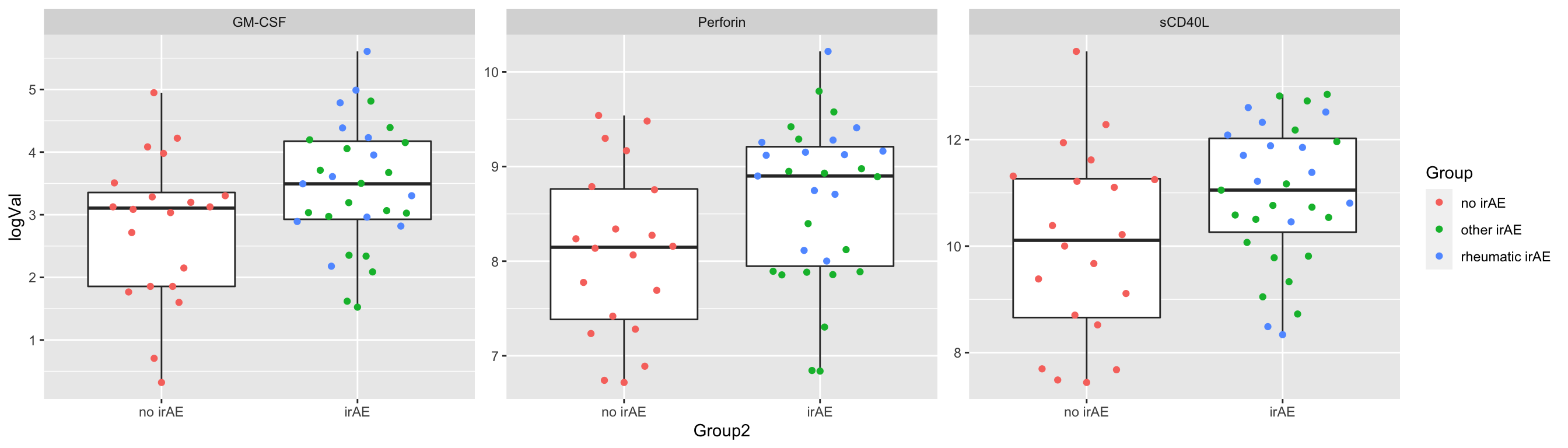
Pair-wise comparison
resPair.followup <- pairwiseT(subTab, "logVal", "Group2", "name")
#filter(resPair.followup, p.value < 0.05)Difference between follow_up and baseline
subTab <- filter(cbaTab, assay == "CBA", condition %in% c("Baseline", "Follow_Up")) %>%
select(name, Group, Group2, condition, logVal, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
mutate(diffVal = Follow_Up - Baseline) %>%
filter(!is.na(diffVal))PCA
subMat <- subTab %>% select(patID, name, diffVal) %>%
pivot_wider(names_from = name, values_from = diffVal) %>%
column_to_rownames("patID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "patID") %>%
left_join(patAnno, by = "patID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC4 0.000210
2 PC20 0.0448 ggplot(pcTab, aes(x=PC20, y=PC4, col = Group2)) +
geom_point()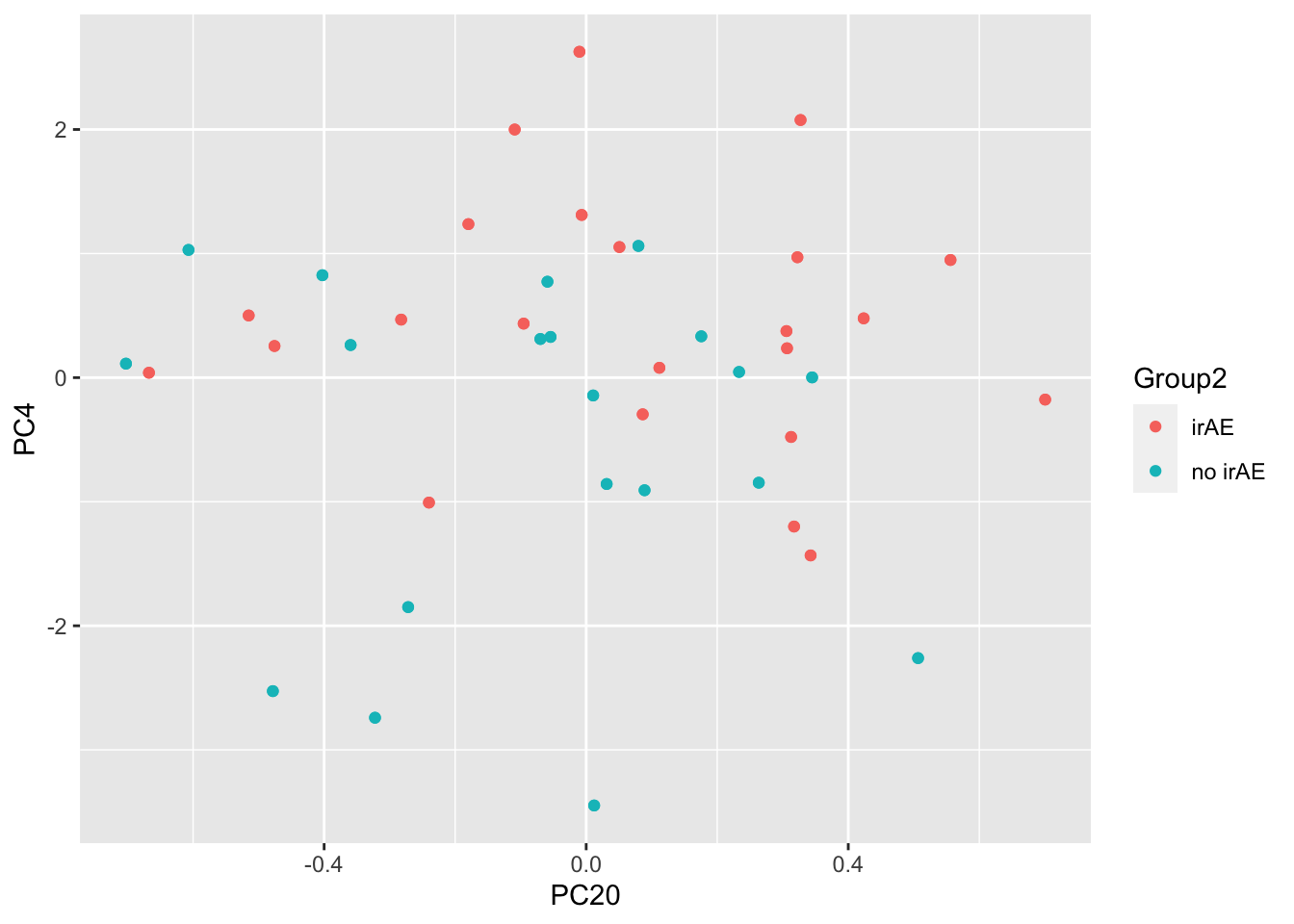
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(diffVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 IL-18 0.00415Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=diffVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")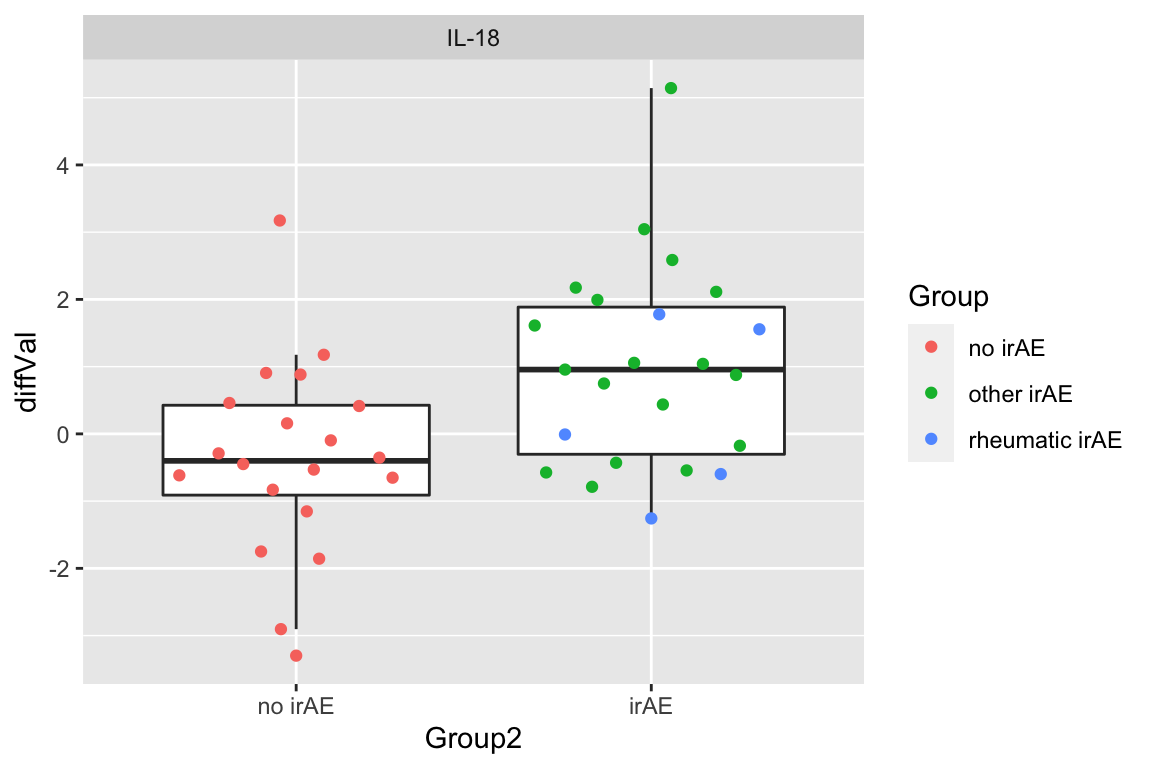
Pair-wise comparison
resPair.diff <- pairwiseT(subTab, "diffVal", "Group2", "name")
#filter(resPair.diff, p.value < 0.05)Summarise the pariwise comparison results using a heatmap
resPair.cba <- bind_rows(
resPair.baseline %>% mutate(condition = "Baseline"),
resPair.followup %>% mutate(condition = "Follow_Up"),
resPair.diff %>% mutate(condition = "Follow_Up - Baseline")
)
plotTab <- mutate(resPair.cba,
comparison = paste0(group2, " ~ ", group1),
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=feature, x=comparison)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
facet_wrap(~condition, ncol=3) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low = "blue", high="red")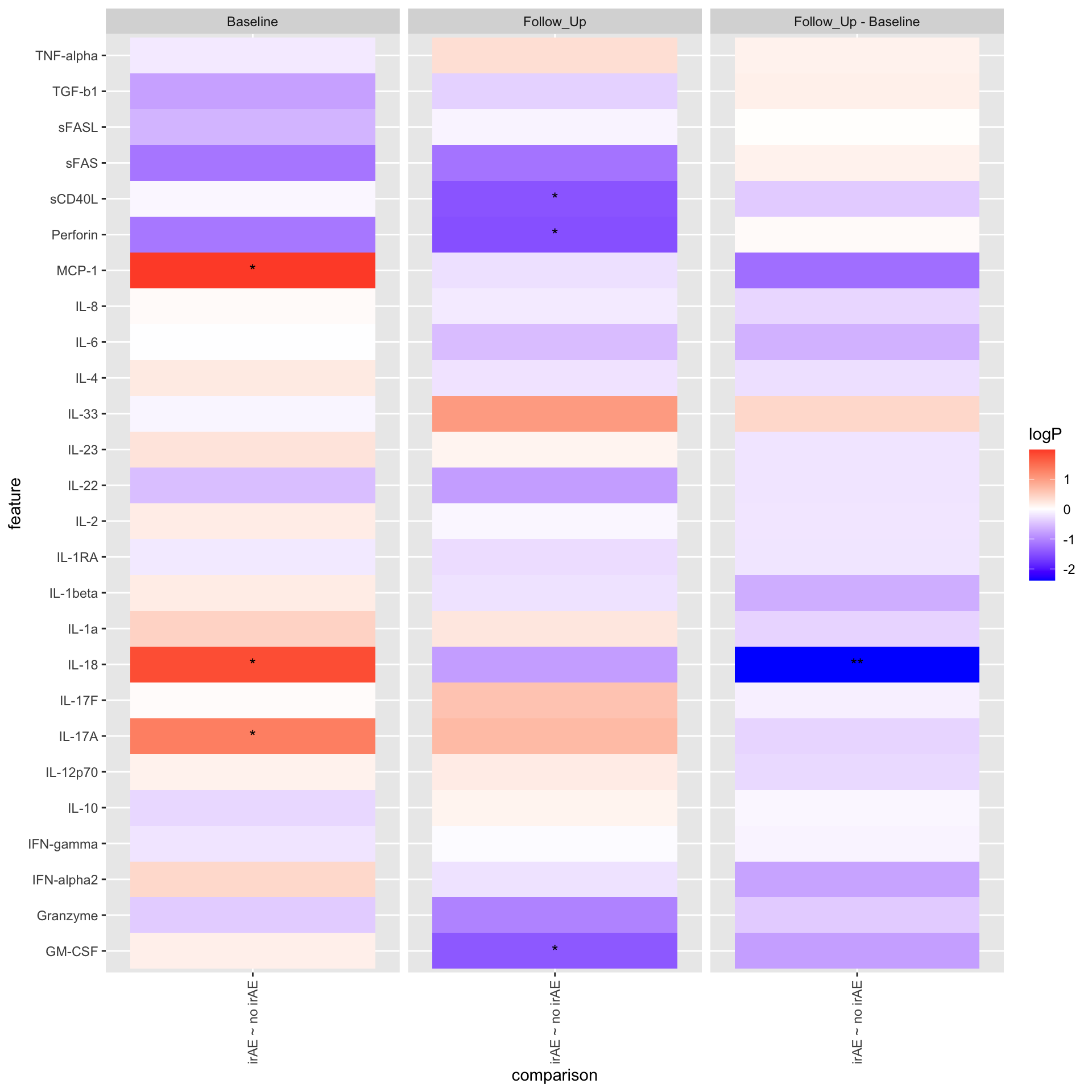
NMR data
nmrTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = value) %>%
mutate(Group2 = ifelse(Group == "no irAE", Group, "irAE")) %>%
mutate(Group2 = factor(Group2, levels = c("no irAE","irAE")))
patAnno <- mutate(patAnno, Group2 = ifelse(Group == "no irAE", Group, "irAE"))Try vsn transformation (not used, may introduce artefact)
nmrMat <- assays(mae)[["nmr"]]
#vsnFit <- vsn::vsnMatrix(nmrMat, minDataPointsPerStratum = 10)
#nmrMat <- predict(vsnFit, nmrMat)
vsnTab <- nmrMat %>% as_tibble(rownames = "name") %>%
pivot_longer(-name, names_to = "sampleID", values_to = "logVal")
#nmrTab <- left_join(nmrTab, vsnTab, by =c("sampleID","name"))Baseline samples
subTab <- filter(nmrTab, condition == "Baseline" )PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 PC11 0.0143
2 PC15 0.0166
3 PC13 0.0475ggplot(pcTab, aes(x=PC11, y=PC15, col = Group2)) +
geom_point()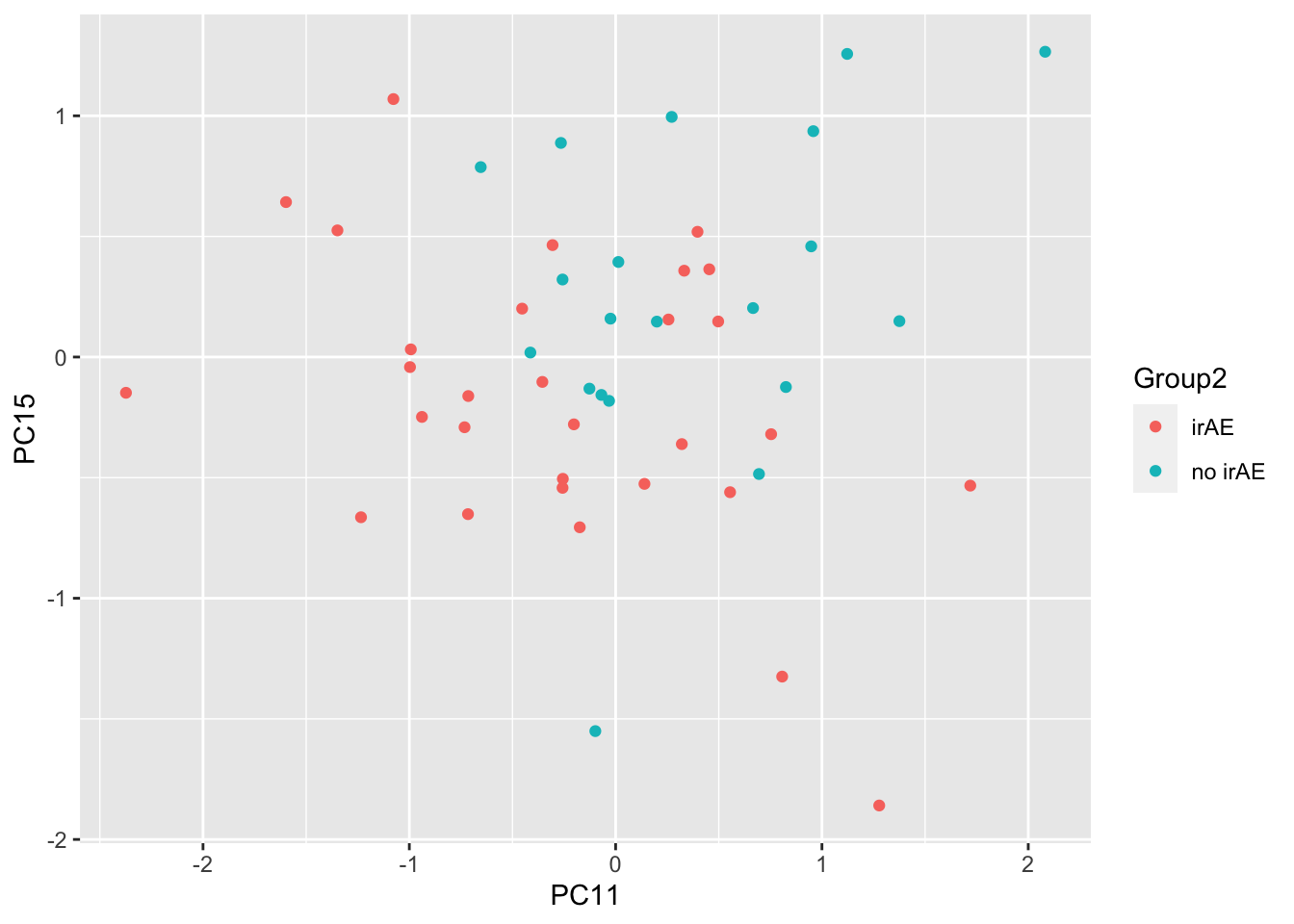
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 Histidine 0.000269
2 Asparagine 0.0379 Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=logVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")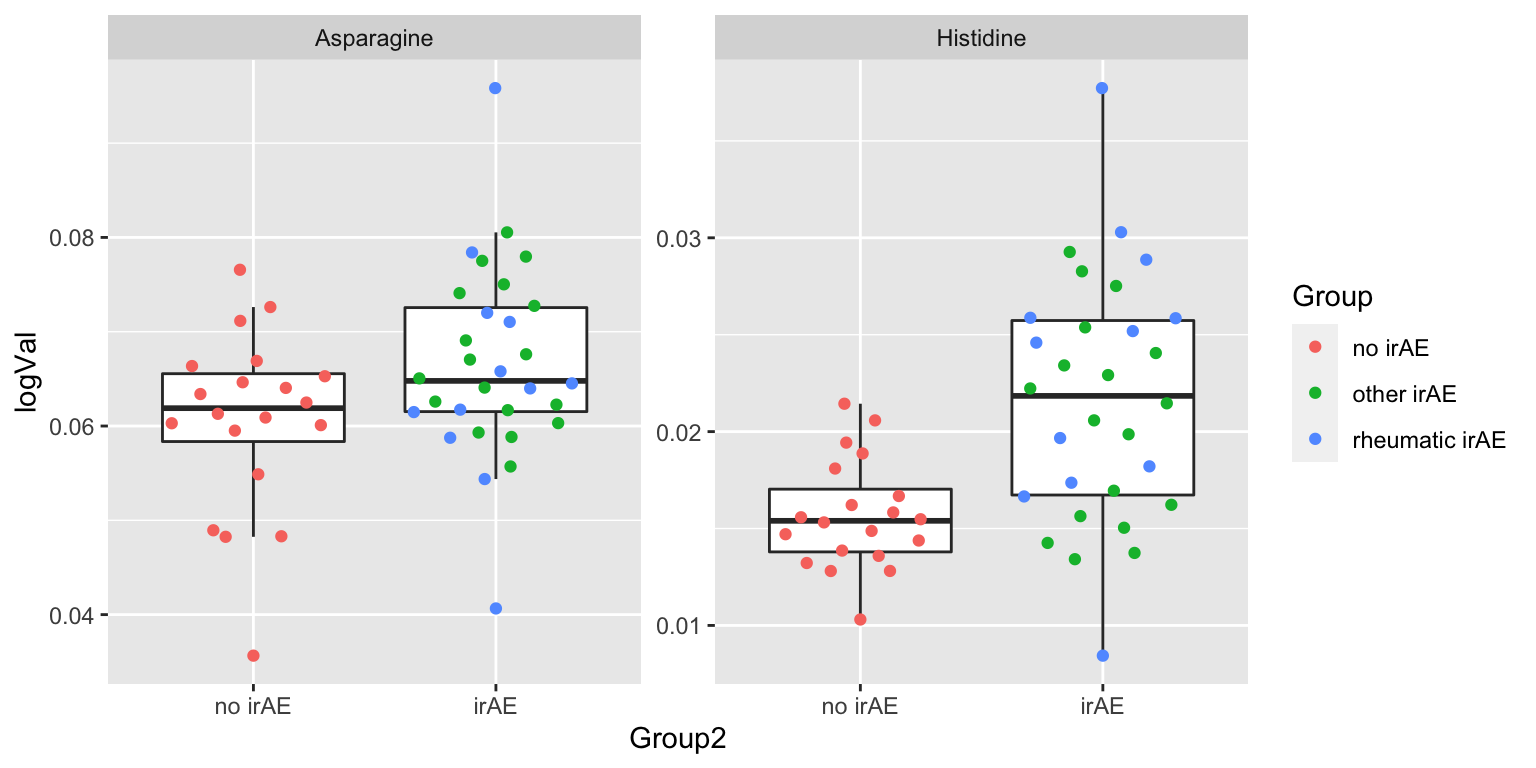
Pair-wise comparison
resPair.baseline <- pairwiseT(subTab, "logVal", "Group2", "name")
#filter(resPair.baseline, p.value < 0.05)Follow-up samples
subTab <- filter(nmrTab, condition == "Follow_Up" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 PC13 0.0111
2 PC16 0.0454ggplot(pcTab, aes(x=PC13, y=PC16, col = Group2)) +
geom_point()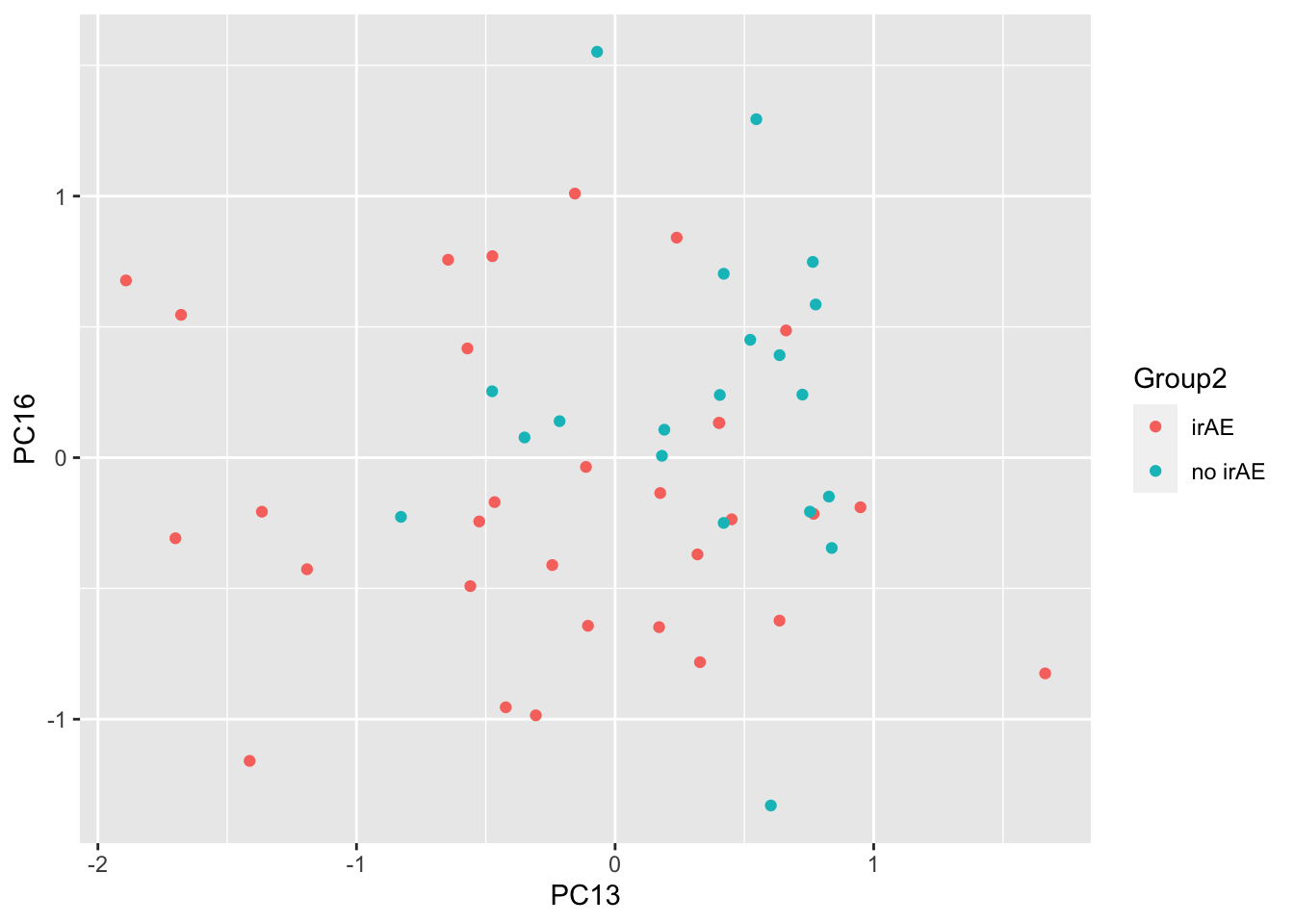
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.1)
resTab.sig# A tibble: 2 × 2
# Groups: name [2]
name p.value
<chr> <dbl>
1 Histidine 0.0513
2 Lactate 0.0827Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=logVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")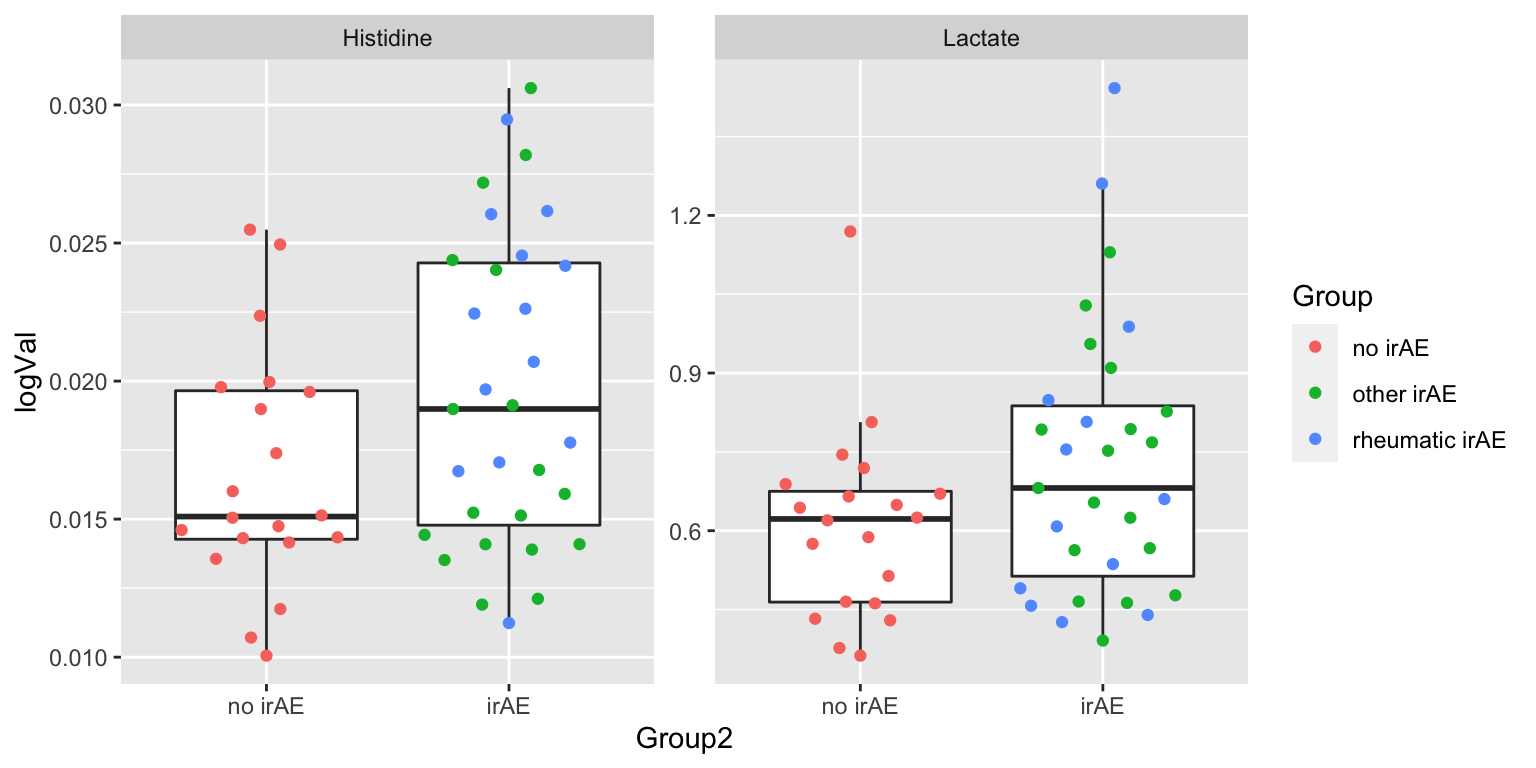
Pair-wise comparison
resPair.followup <- pairwiseT(subTab, "logVal", "Group2", "name")Use difference between follow_up and baseline
subTab <- filter(nmrTab, condition %in% c("Baseline", "Follow_Up")) %>%
select(name, Group, condition, Group2,logVal, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
mutate(diffVal = Follow_Up - Baseline) %>%
filter(!is.na(diffVal))PCA
subMat <- subTab %>% select(patID, name, diffVal) %>%
pivot_wider(names_from = name, values_from = diffVal) %>%
column_to_rownames("patID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "patID") %>%
left_join(patAnno, by = "patID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group2)) +
geom_point()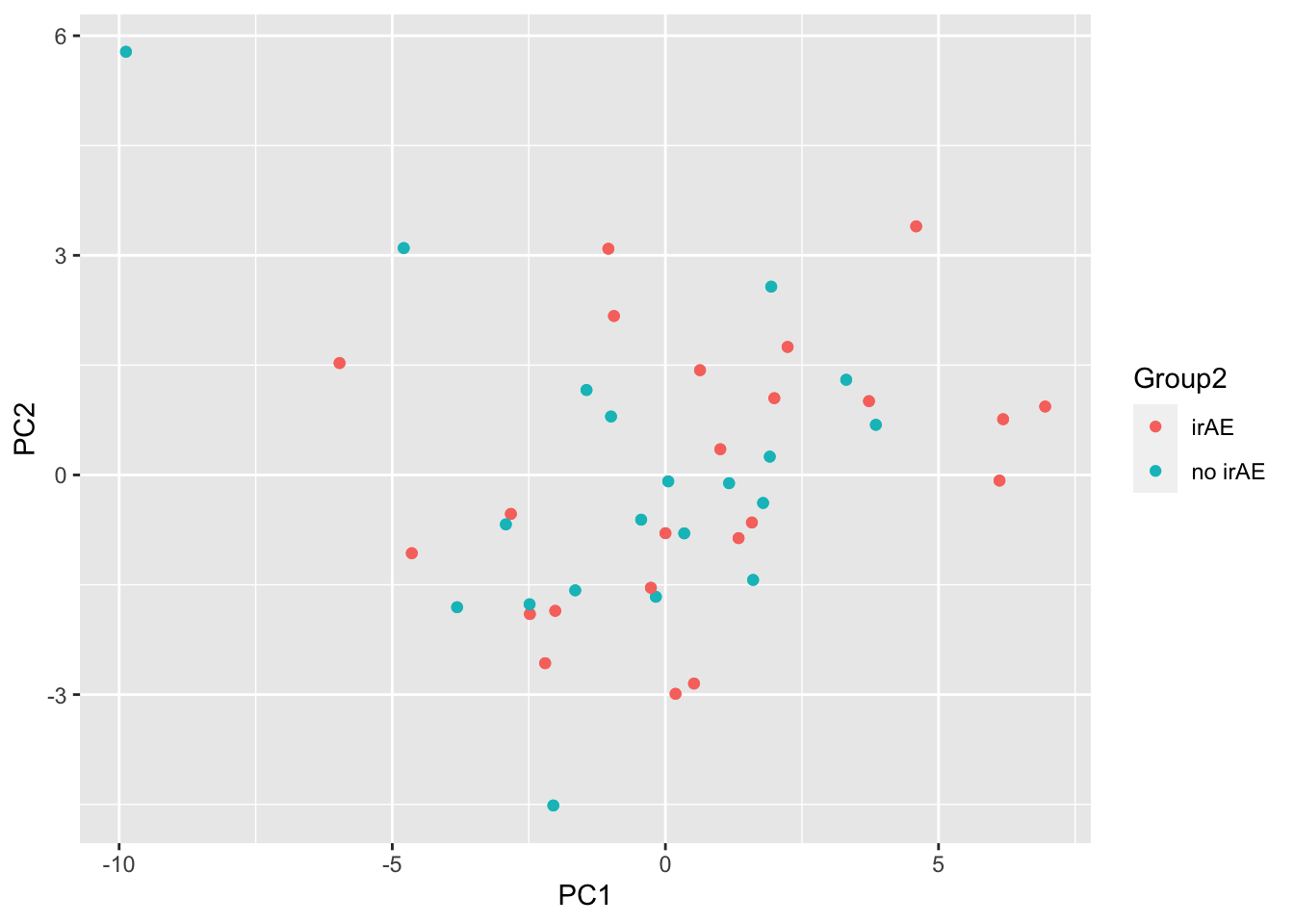
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group2)) +
geom_point()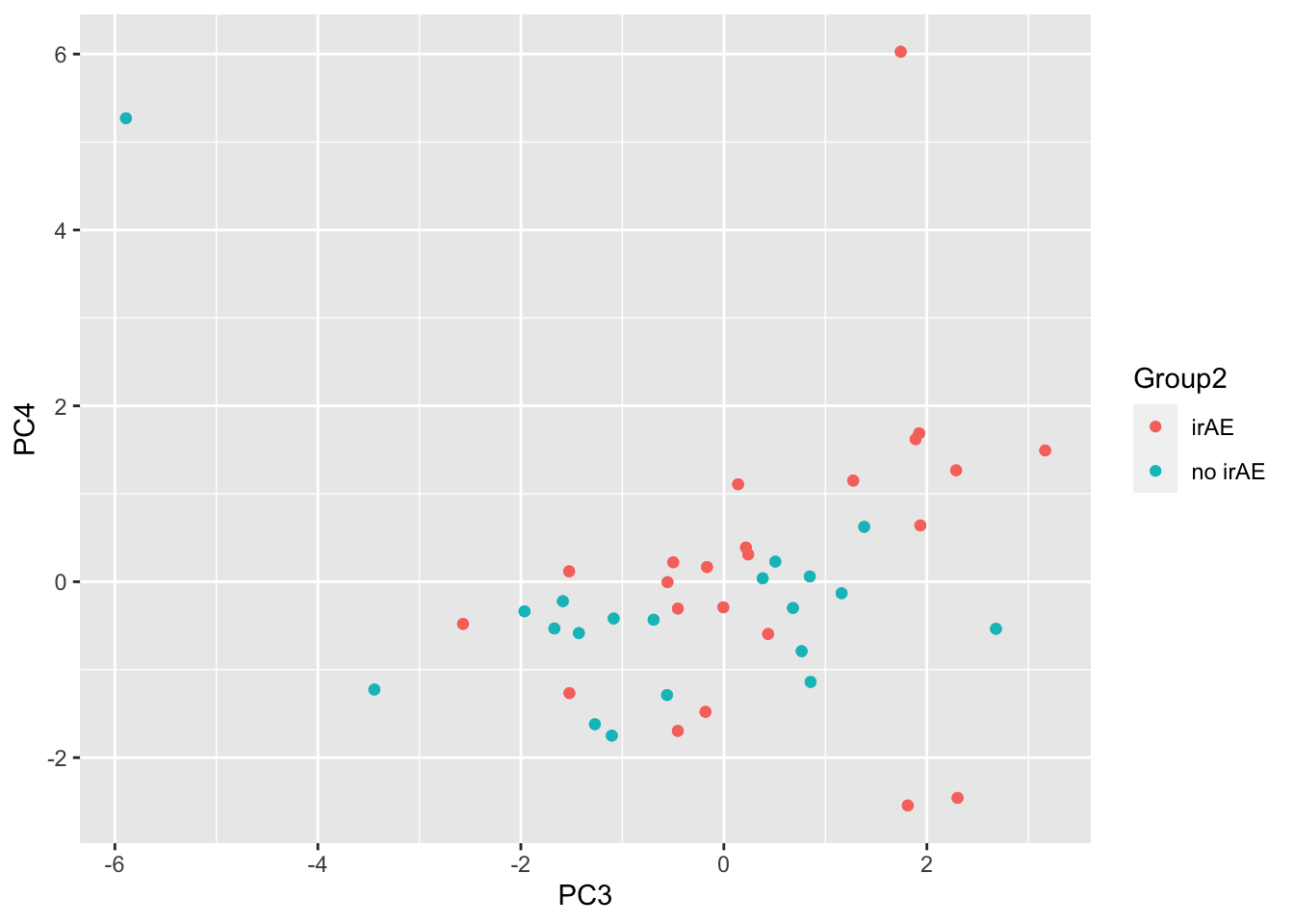
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group2) %>%
pivot_longer(-c(sampleID, Group2))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 PC3 0.00379
2 PC20 0.0179
3 PC12 0.0201 ggplot(pcTab, aes(x=PC3, y=PC20, col = Group2)) +
geom_point()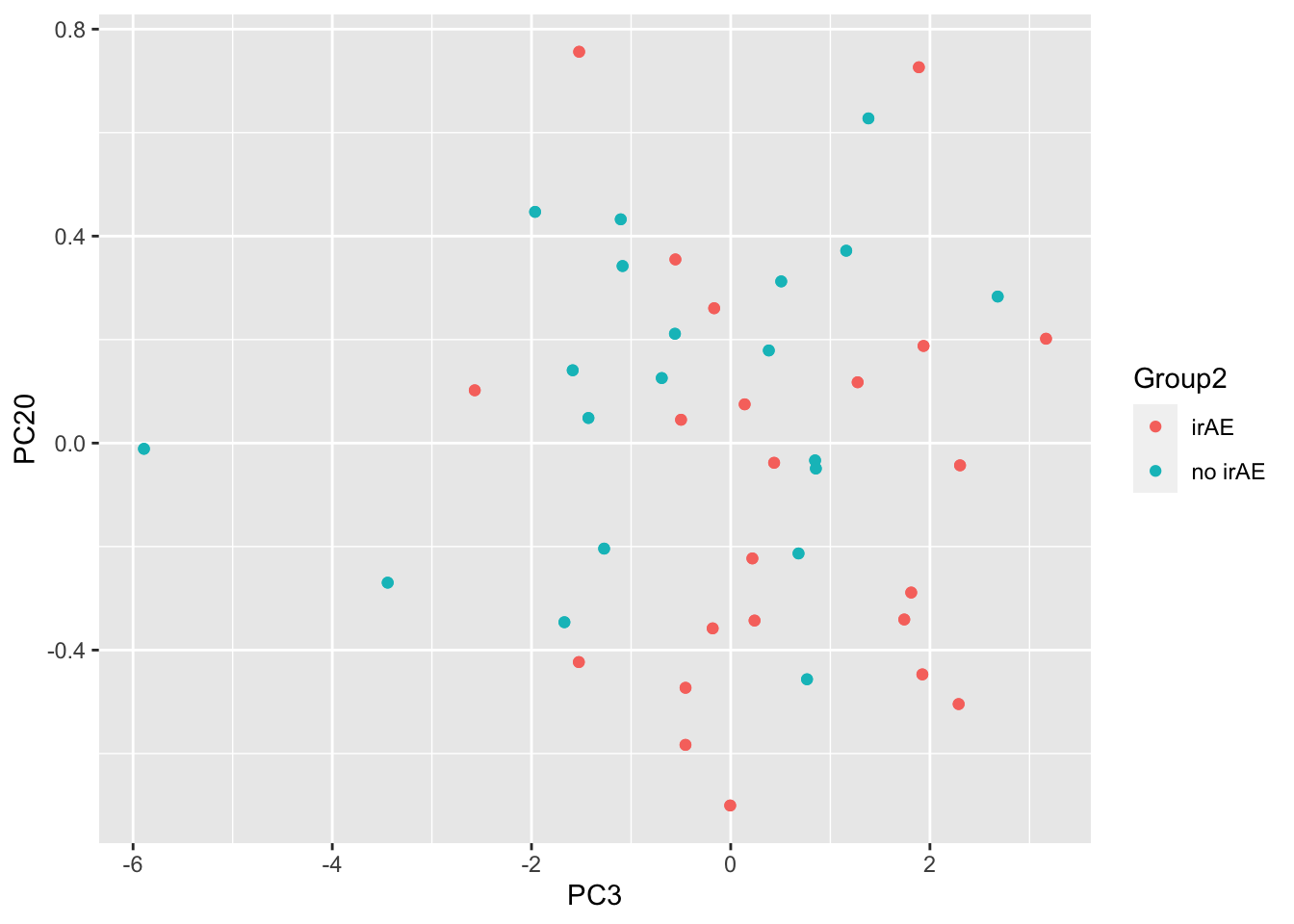
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(diffVal ~ Group2,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group2") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 Choline 0.00564
2 Asparagine 0.0298
3 Histidine 0.0303 Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group2, y=diffVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col=Group)) +
facet_wrap(~name, scales = "free")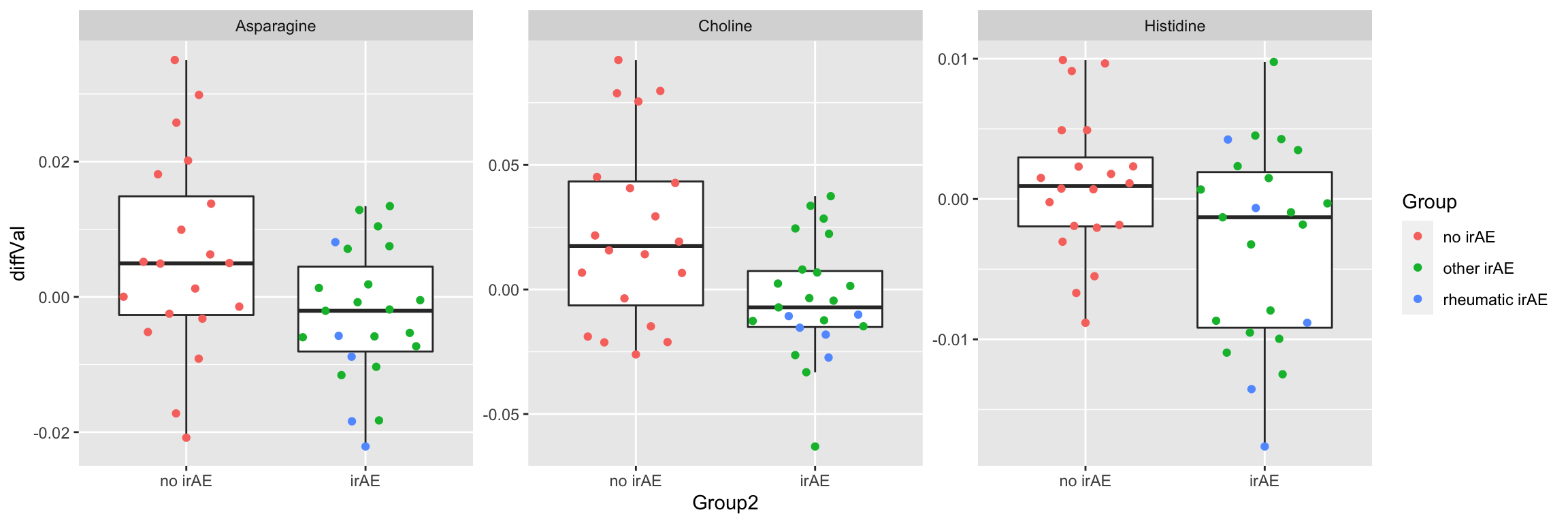
Pair-wise comparison
resPair.diff <- pairwiseT(subTab, "diffVal", "Group2", "name")Summarise the pariwise comparison results using a heatmap
resPair.cba <- bind_rows(
resPair.baseline %>% mutate(condition = "Baseline"),
resPair.followup %>% mutate(condition = "Follow_Up"),
resPair.diff %>% mutate(condition = "Follow_Up - Baseline")
)
plotTab <- mutate(resPair.cba,
comparison = paste0(group2, " ~ ", group1),
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=feature, x=comparison)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
facet_wrap(~condition, ncol=3) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low="blue", high="red")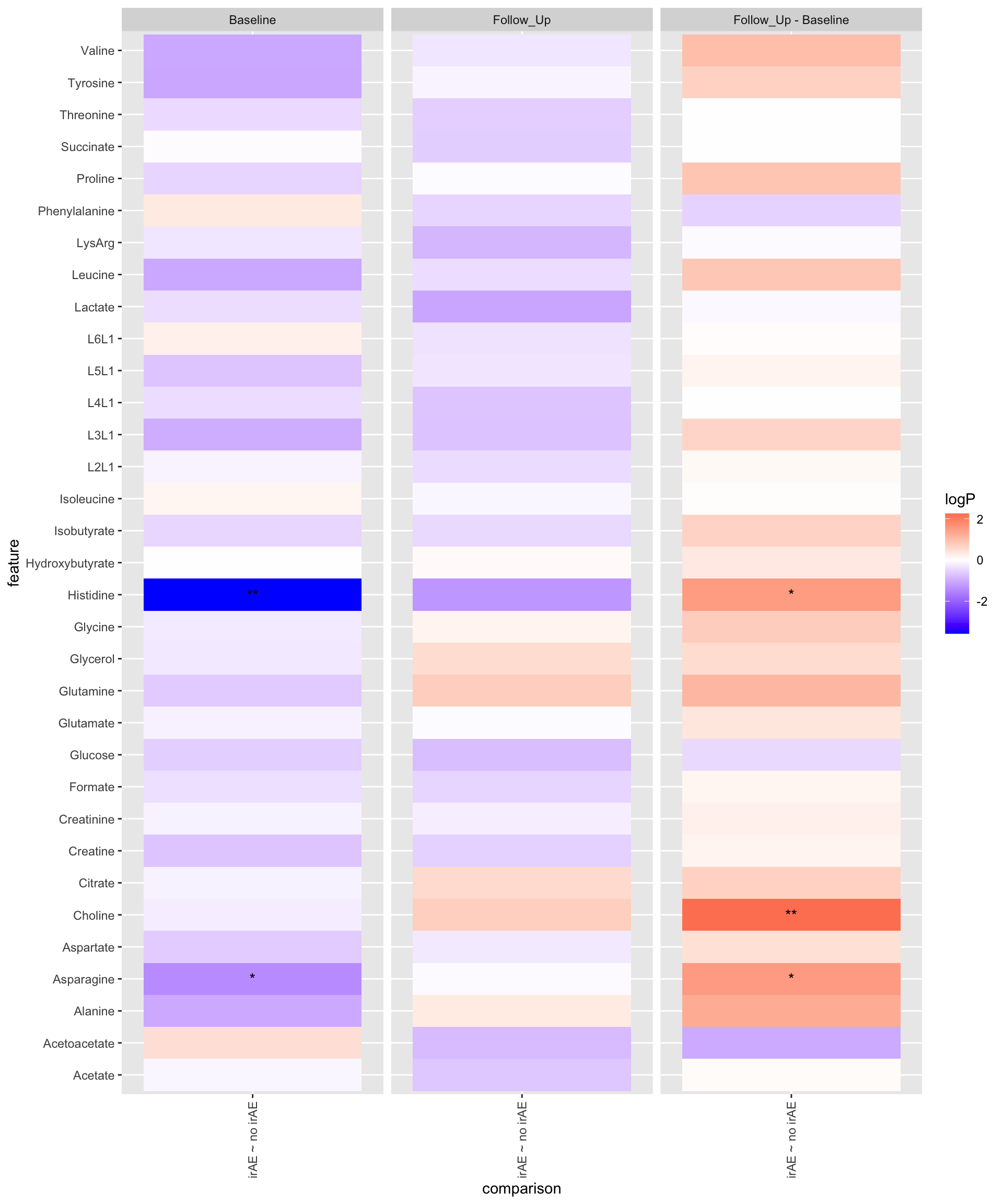
Analysis focused on none-cancer patients
CBA data
cbaTab <- filter(fullTab, assay == "CBA") %>%
mutate(logVal = glog2(value))Baseline samples
subTab <- filter(cbaTab, condition == "noMalignancy" ) PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group, shape = factor(dateOfAcquisition_CBA))) +
geom_point()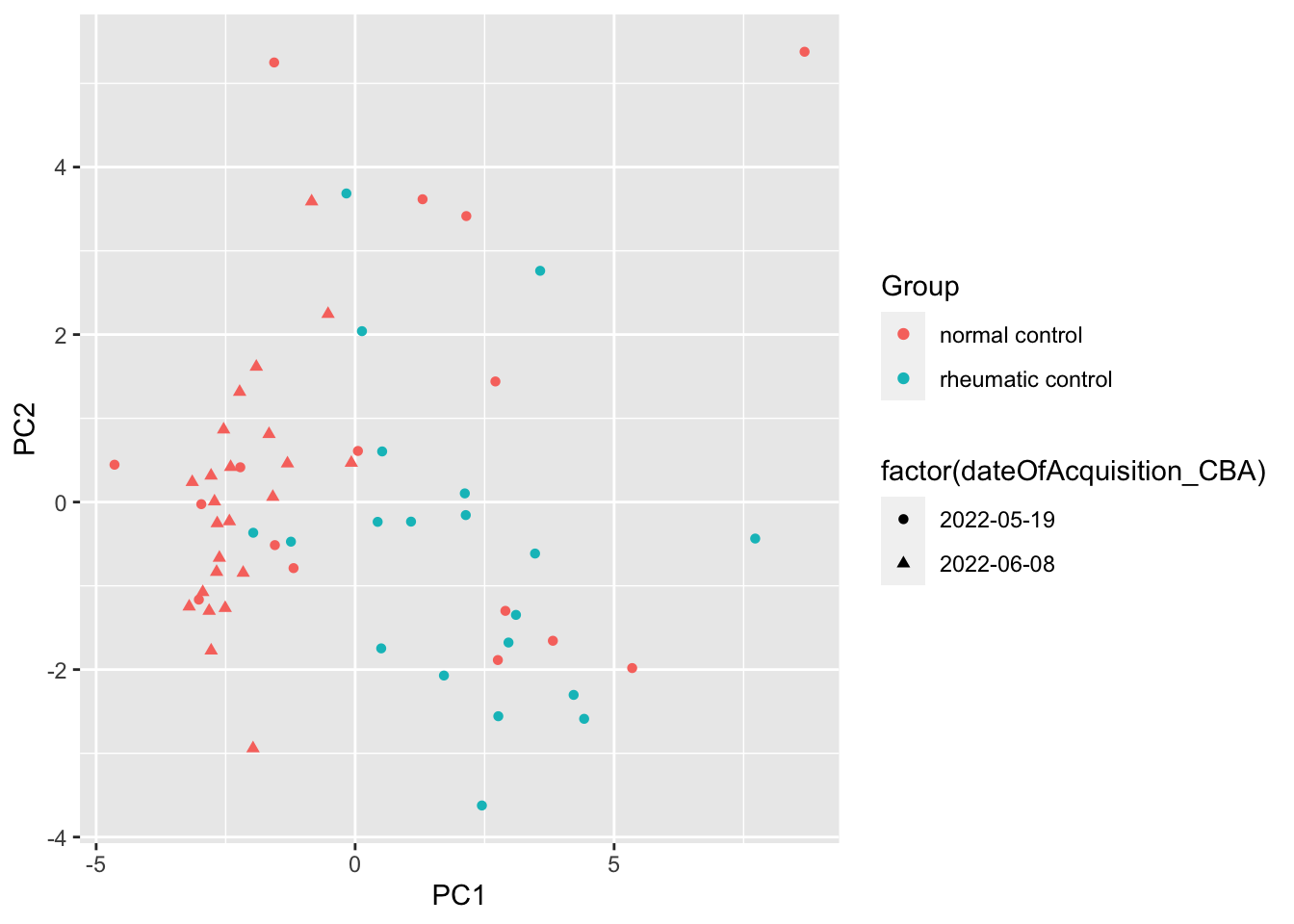
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group, shape = factor(dateOfAcquisition_CBA))) +
geom_point()
Batch can be a confounder, remove sample with the second batch
subTab <- filter(subTab, as.character(dateOfAcquisition_CBA) != "2022-06-08")
subMat <- subTab %>%
select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group, shape = factor(dateOfAcquisition_CBA))) +
geom_point()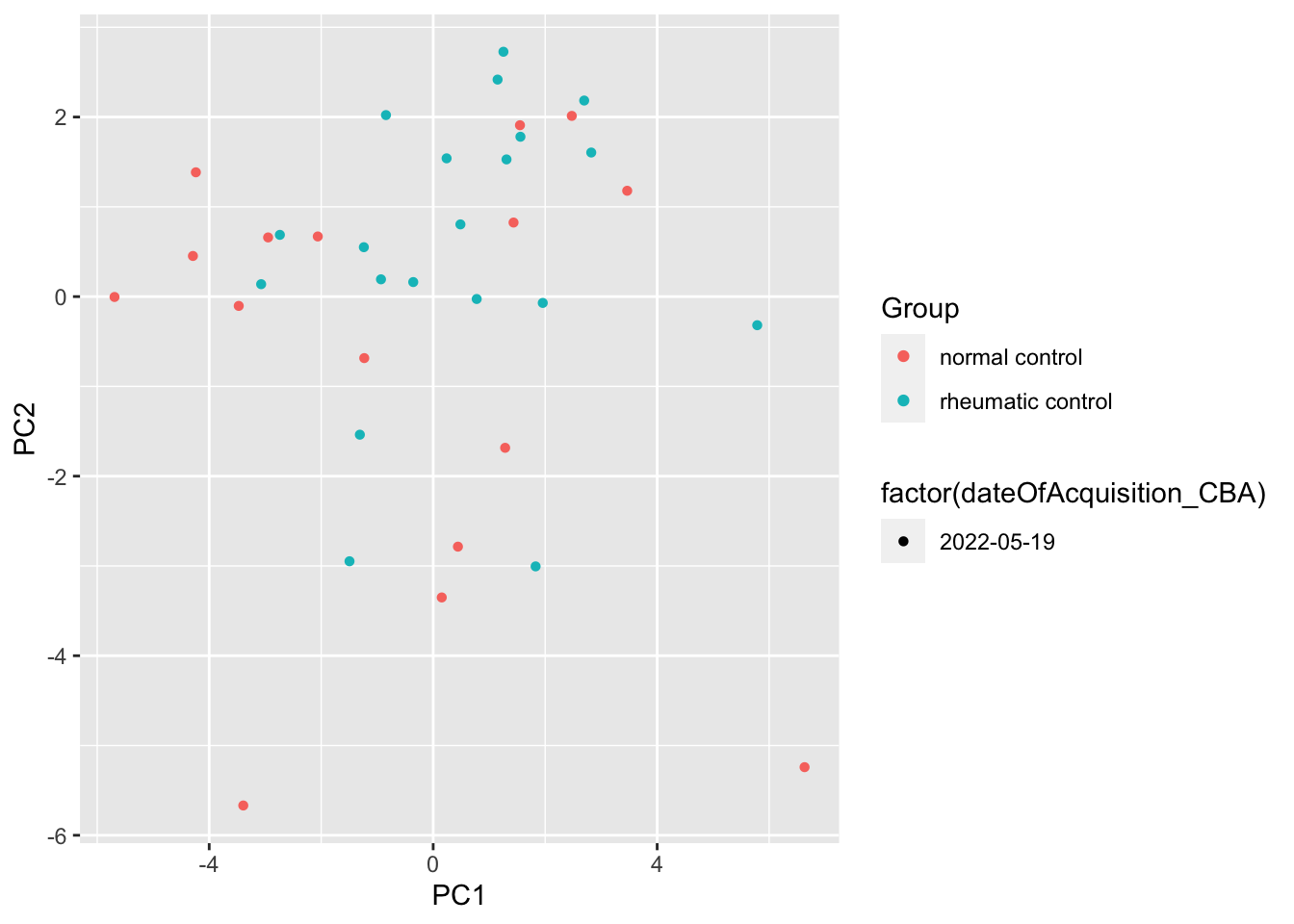
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group, shape = factor(dateOfAcquisition_CBA))) +
geom_point()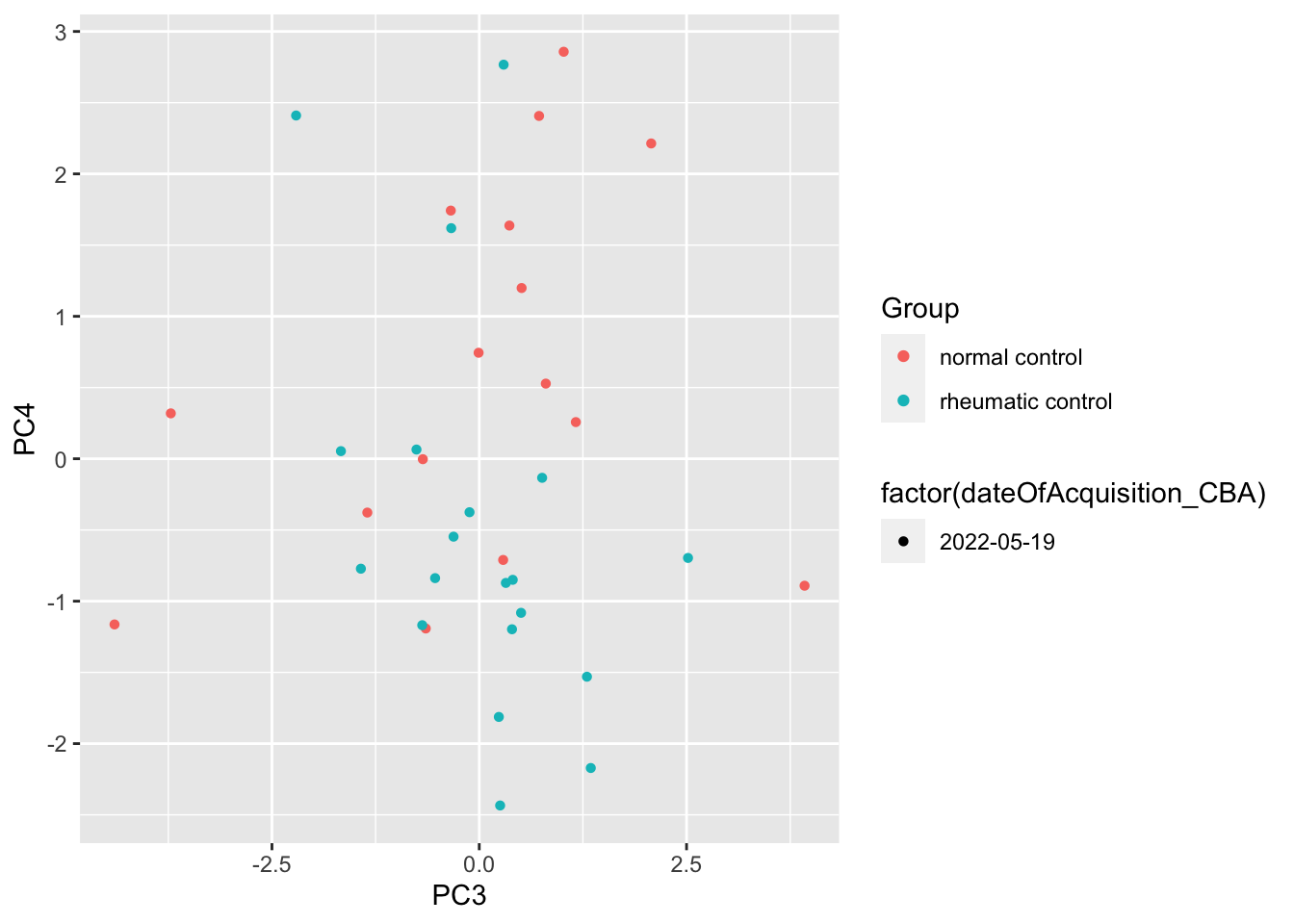
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 PC4 0.0222ggplot(pcTab, aes(x=PC1, y=PC4, col = Group, shape = factor(dateOfAcquisition_CBA))) +
geom_point()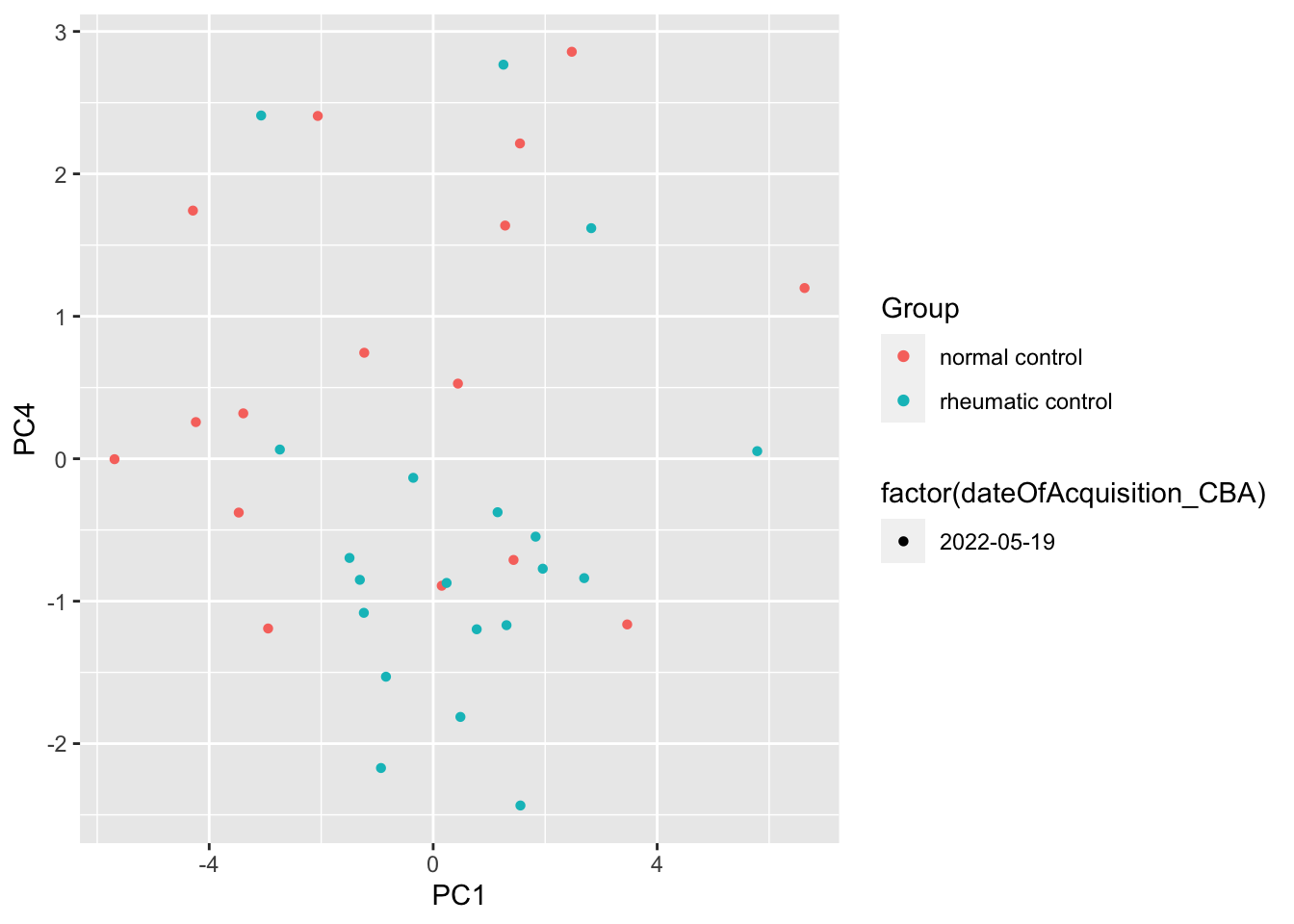
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~car::Anova(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 IL-12p70 0.0317
2 sCD40L 0.0456
3 IL-33 0.0458Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot(outlier.shape = NA) + ggbeeswarm::geom_quasirandom(aes(col = Group)) +
facet_wrap(~name, scales = "free")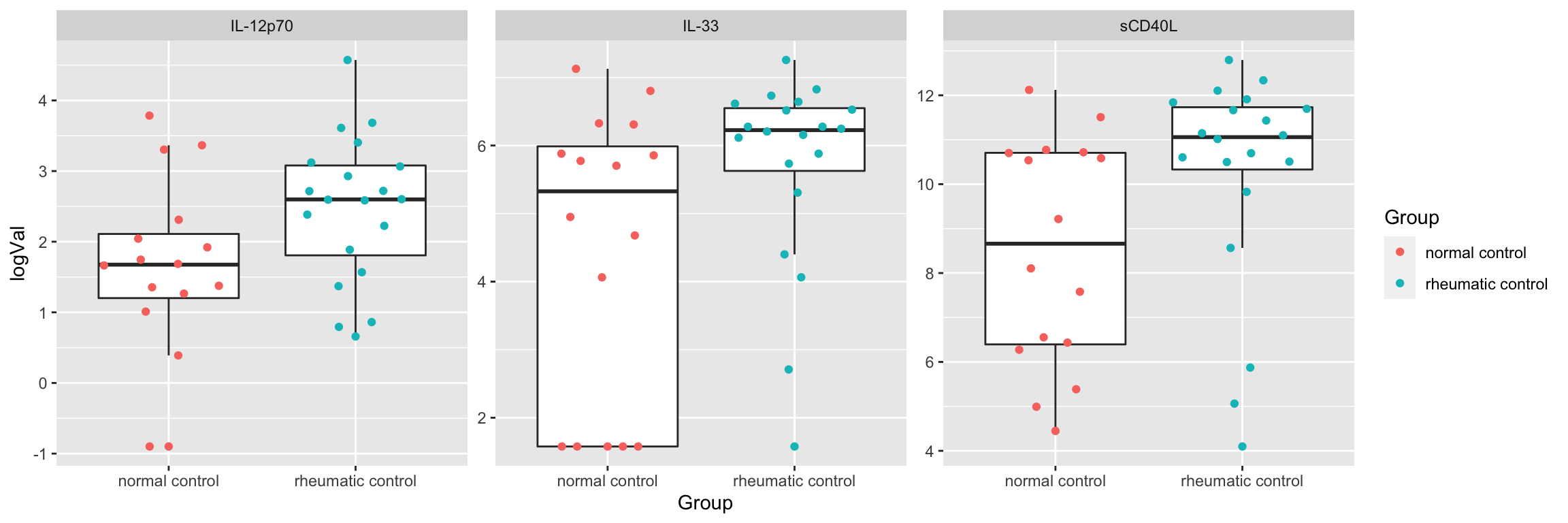
NMR data
nmrTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = value)Try vsn transformation
nmrMat <- assays(mae)[["nmr"]]
#vsnFit <- vsn::vsnMatrix(nmrMat, minDataPointsPerStratum = 10)
#nmrMat <- predict(vsnFit, nmrMat)
vsnTab <- nmrMat %>% as_tibble(rownames = "name") %>%
pivot_longer(-name, names_to = "sampleID", values_to = "logVal")
#nmrTab <- left_join(nmrTab, vsnTab, by =c("sampleID","name"))Baseline samples
subTab <- filter(nmrTab, condition == "noMalignancy" )PCA
subMat <- subTab %>% select(sampleID, name, logVal) %>%
pivot_wider(names_from = name, values_from = logVal) %>%
column_to_rownames("sampleID") %>% as.matrix()
pcRes <- prcomp(subMat, center = TRUE, scale. = TRUE)
pcTab <- pcRes$x %>% as_tibble(rownames = "sampleID") %>%
left_join(patAnno, by = "sampleID")
ggplot(pcTab, aes(x=PC1, y=PC2, col = Group)) +
geom_point()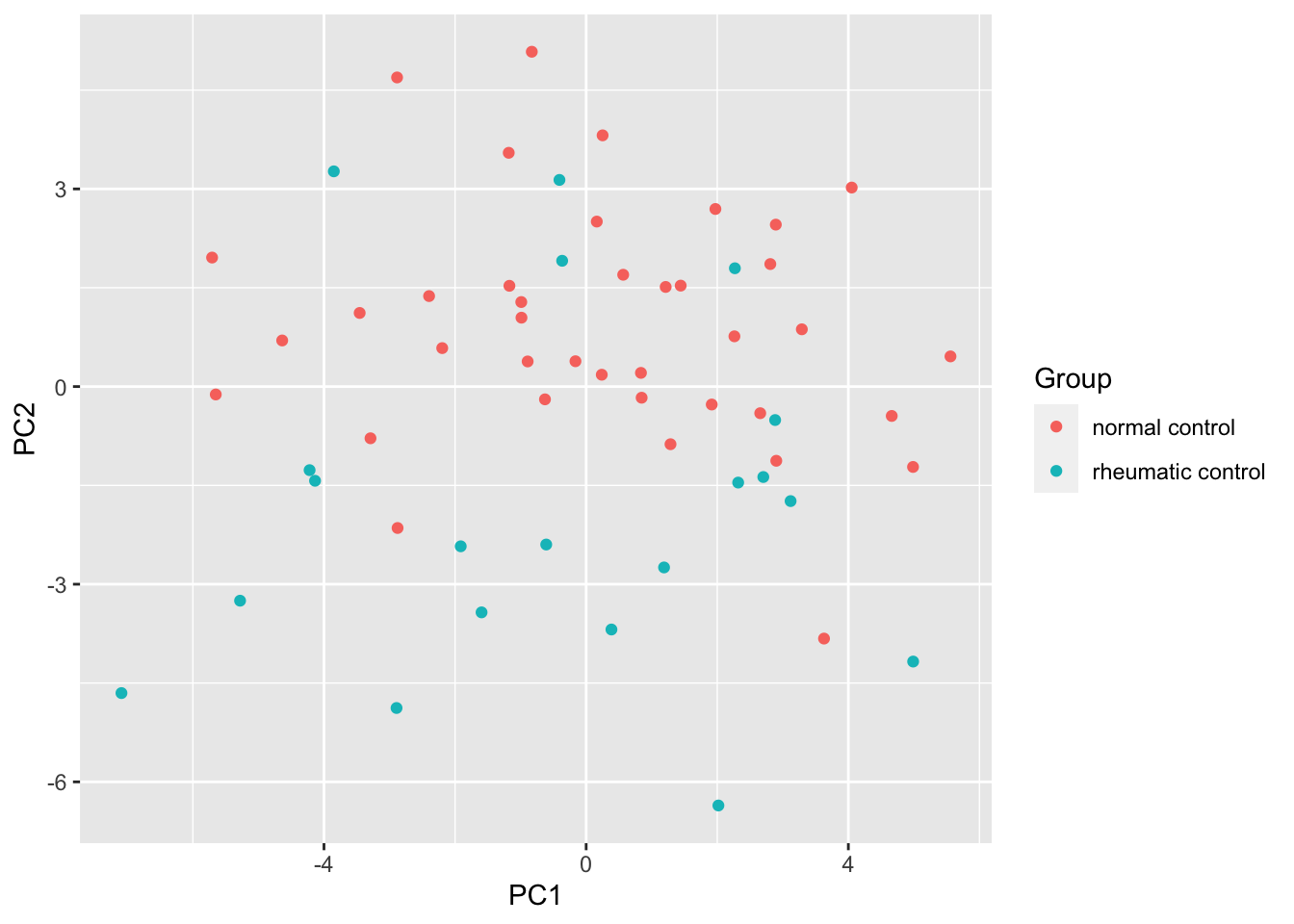
ggplot(pcTab, aes(x=PC3, y=PC4, col = Group)) +
geom_point()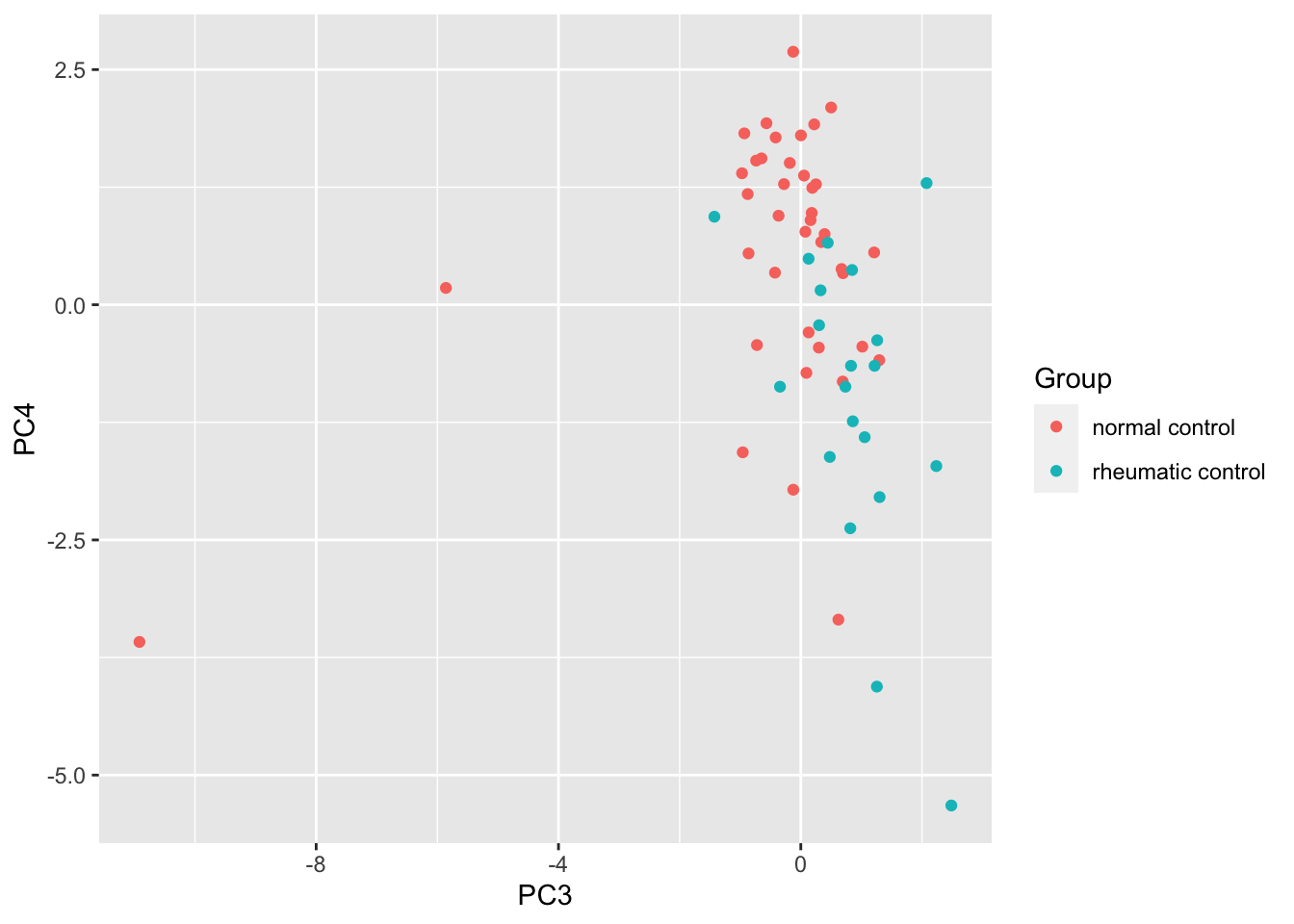
Any PC separate different group?
testTab <- pcTab %>% select(sampleID, PC1:PC20, Group) %>%
pivot_longer(-c(sampleID, Group))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(value ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>% arrange(p.value)
resTab %>% select(name, p.value) %>% filter(p.value <=0.05)# A tibble: 3 × 2
# Groups: name [3]
name p.value
<chr> <dbl>
1 PC2 0.0000203
2 PC4 0.000605
3 PC3 0.0103 ggplot(pcTab, aes(x=PC2, y=PC4, col = Group)) +
geom_point()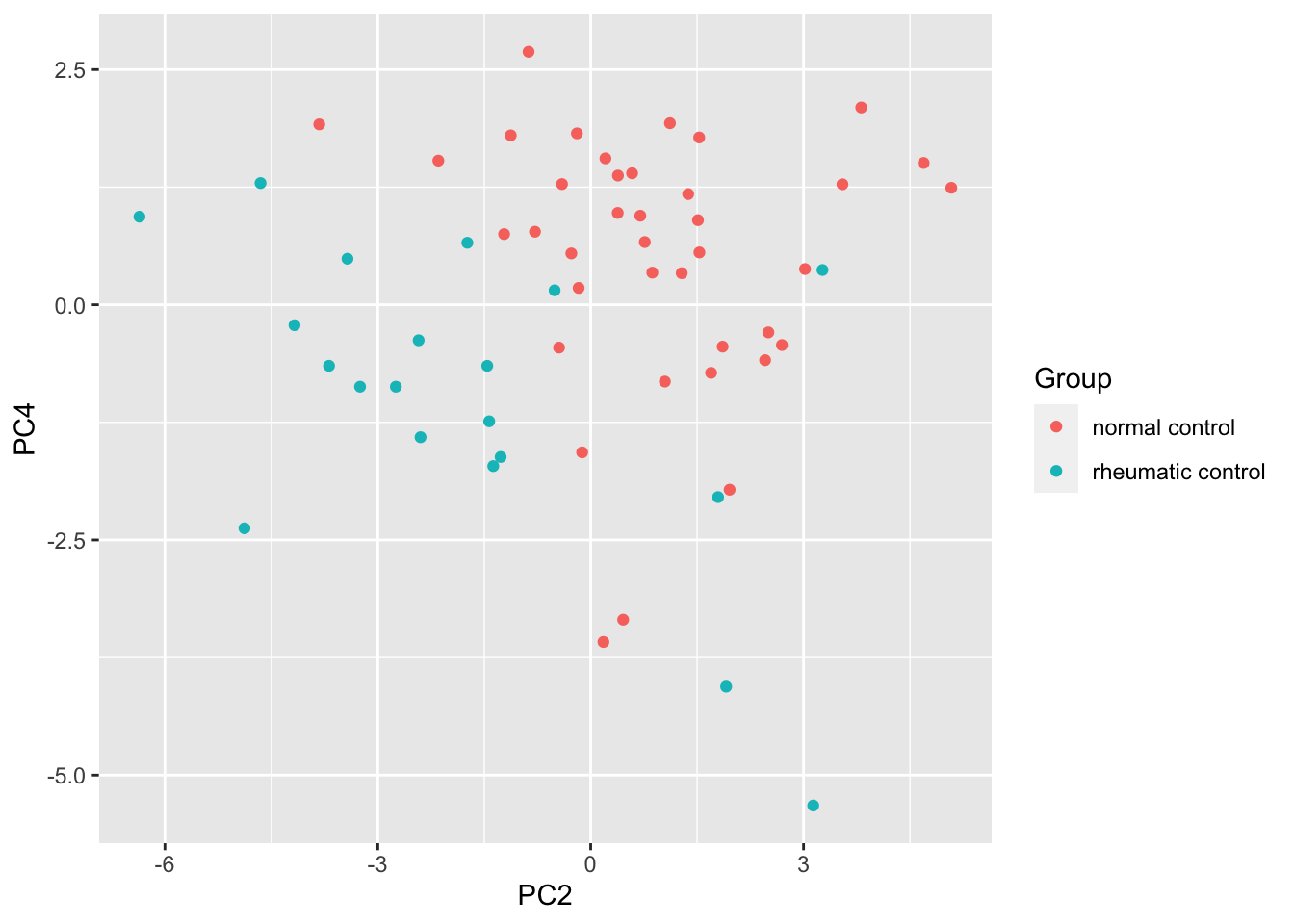
ANOVA
resTab <- subTab %>% group_by(name) %>% nest() %>%
mutate(m = map(data, ~aov(lm(logVal ~ Group,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
filter(term == "Group") %>%
arrange(p.value) %>%
select(name, p.value)
resTab.sig <- filter(resTab, p.value < 0.05)
resTab.sig# A tibble: 9 × 2
# Groups: name [9]
name p.value
<chr> <dbl>
1 Asparagine 0.00000000158
2 Histidine 0.0000000241
3 L2L1 0.00000152
4 L6L1 0.00000904
5 Aspartate 0.0000500
6 Lactate 0.000381
7 Creatine 0.000594
8 L4L1 0.00429
9 Hydroxybutyrate 0.0125 Plot
plotTab <- filter(subTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot() + geom_point() +
facet_wrap(~name, scales = "free")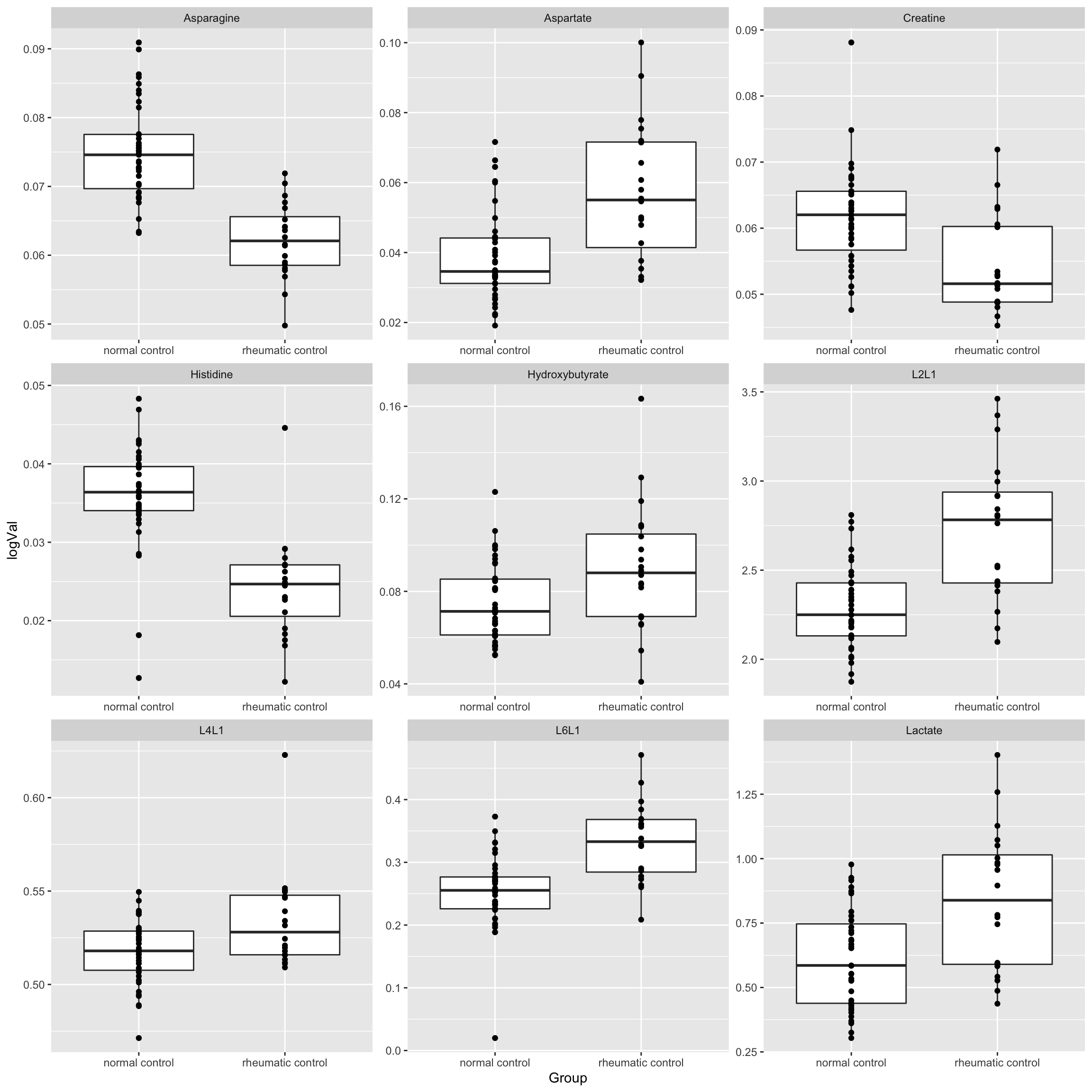
Compare follow-up sample to baseline samples in cancer patients
CBA data
cbaTab <- filter(fullTab, assay == "CBA") %>%
mutate(logVal = glog2(value)) %>%
mutate(Group = factor(Group, levels = c("no irAE","rheumatic irAE","other irAE"))) %>%
filter(Group != "noMalignancy")testTab <- cbaTab %>% select(name, logVal, condition, Group, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
filter(!is.na(Baseline), !is.na(Follow_Up))
resTab <- group_by(testTab, name, Group) %>% nest() %>%
mutate(m = map(data, ~t.test(.$Baseline, .$Follow_Up, paired = TRUE))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name,Group, estimate, p.value) %>%
arrange(p.value)P-value heatmap
plotTab <- mutate(resTab,
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=Group, x=name)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low="blue", high="red")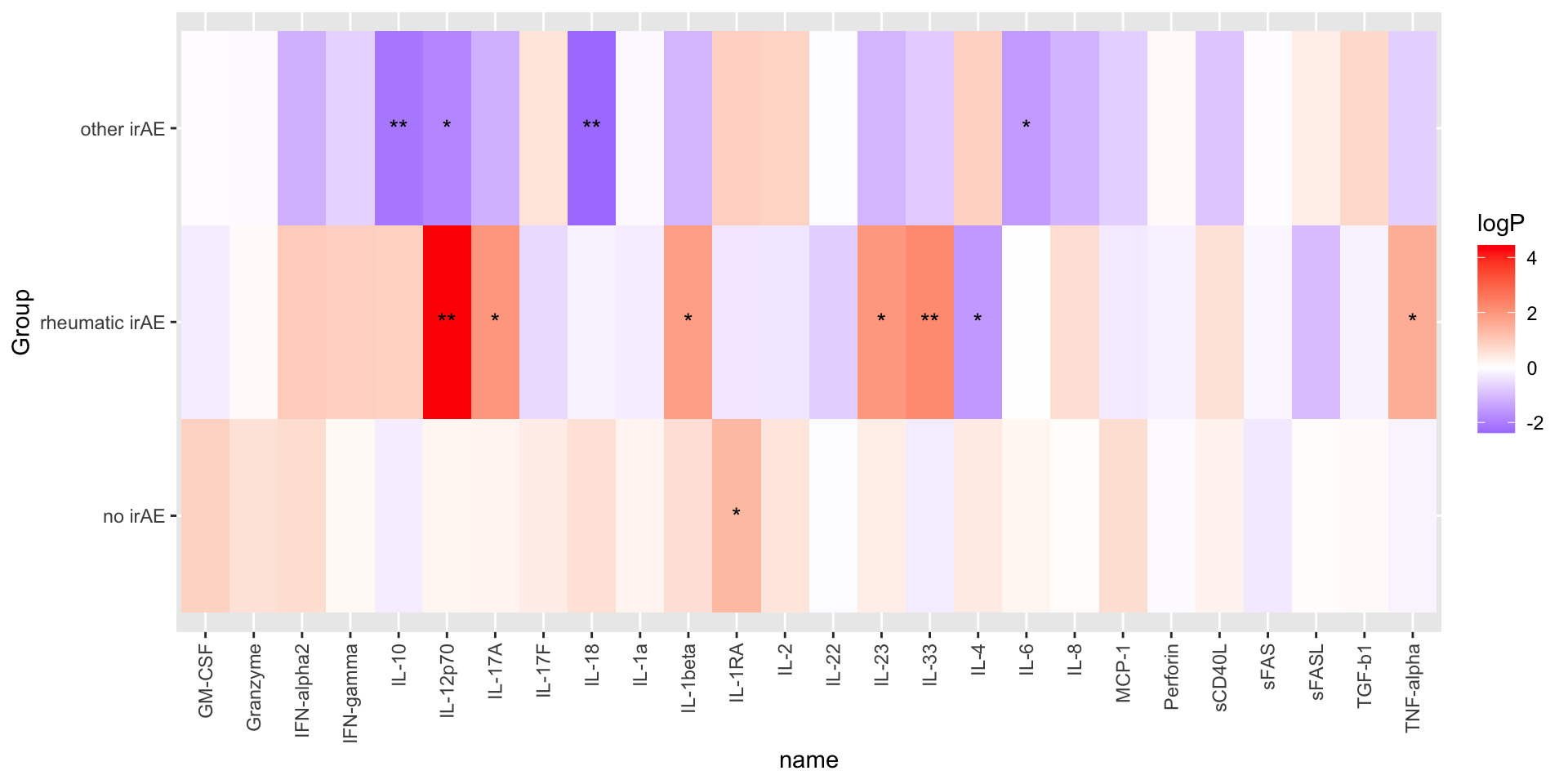
Plot individual associations
resTab.sig <- filter(resTab, p.value <=0.05)
pList <- lapply(unique(resTab.sig$name), function(nn) {
pTab <- filter(resTab, name ==nn)
plotTab <- filter(cbaTab, name ==nn) %>%
left_join(pTab, by = c("name","Group")) %>%
mutate(title = sprintf("%s (%s)", Group, formatC(p.value, digits = 2)))
ggplot(plotTab, aes(x=condition, y=logVal, col = title)) +
geom_point() +
geom_line(aes(group=patID), linetype = "dotted") +
facet_wrap(~title) + ggtitle(nn) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "none")
})
cowplot::plot_grid(plotlist = pList, ncol=2)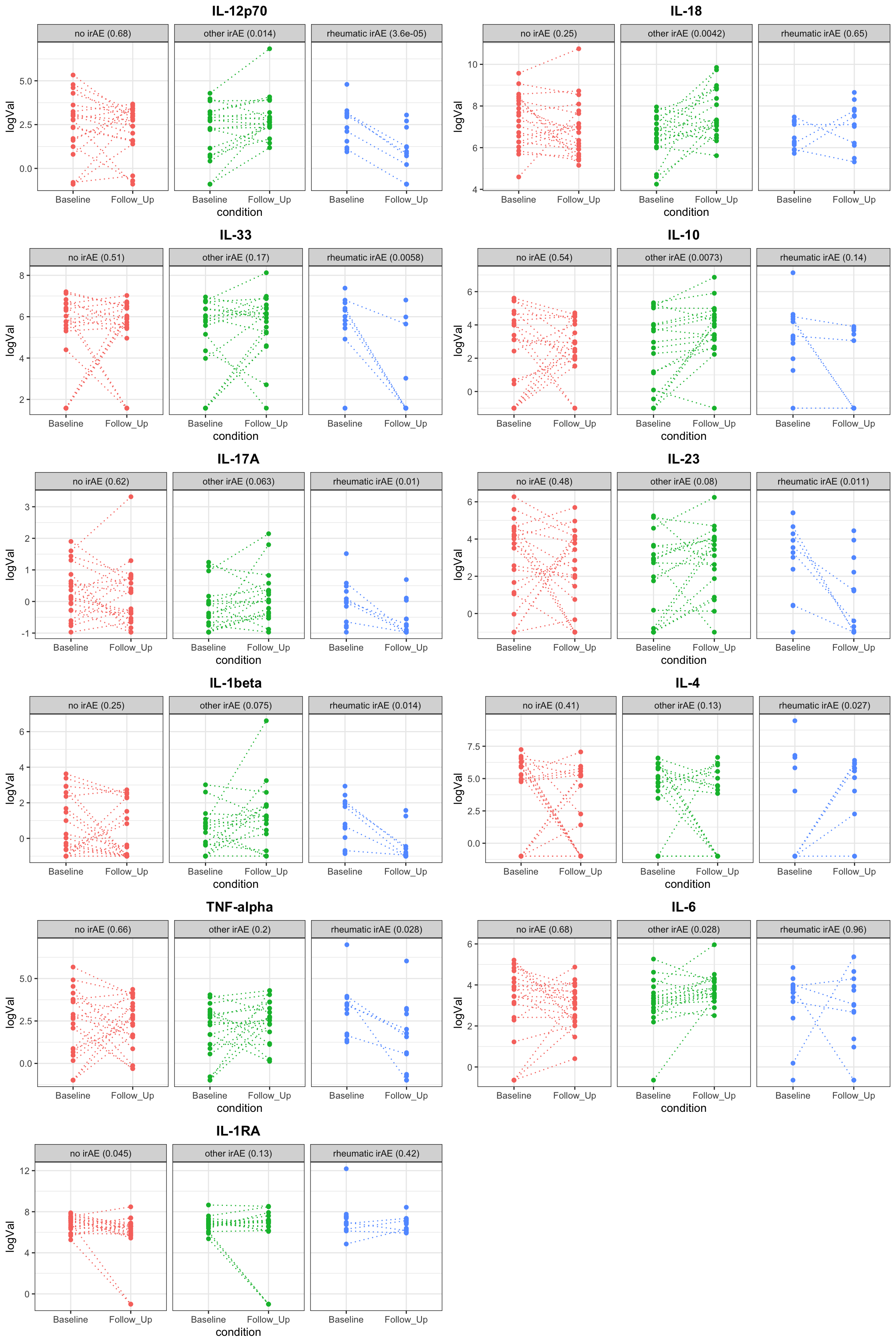
NMR data
cbaTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = value) %>%
mutate(Group = factor(Group, levels = c("no irAE","rheumatic irAE","other irAE"))) %>%
filter(Group != "noMalignancy")testTab <- cbaTab %>% select(name, logVal, condition, Group, patID) %>%
pivot_wider(names_from = condition, values_from = logVal) %>%
filter(!is.na(Baseline), !is.na(Follow_Up))
resTab <- group_by(testTab, name, Group) %>% nest() %>%
mutate(m = map(data, ~t.test(.$Baseline, .$Follow_Up, paired = TRUE))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name,Group, estimate, p.value) %>%
arrange(p.value)P-value heatmap
plotTab <- mutate(resTab,
logP = -log10(p.value)*sign(estimate),
star = case_when(
p.value <= 0.01 ~ "**",
p.value <= 0.05 ~ "*",
TRUE ~ ""
))
ggplot(plotTab, aes(y=Group, x=name)) +
geom_tile(aes(fill = logP)) +
geom_text(aes(label = star)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) +
scale_fill_gradient2(low="blue", high="red")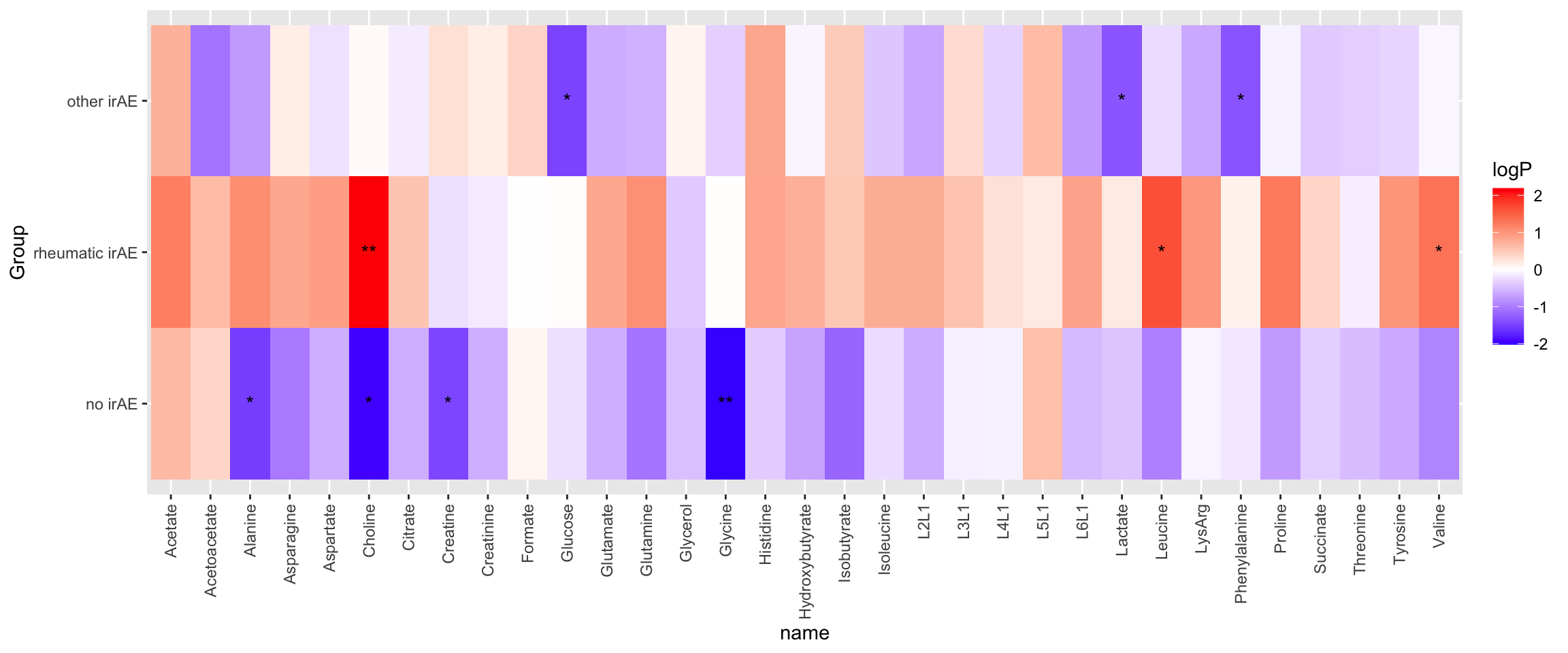
Plot individual associations
resTab.sig <- filter(resTab, p.value <=0.05)
pList <- lapply(unique(resTab.sig$name), function(nn) {
pTab <- filter(resTab, name ==nn)
plotTab <- filter(cbaTab, name ==nn) %>%
left_join(pTab, by = c("name","Group")) %>%
mutate(title = sprintf("%s (%s)", Group, formatC(p.value, digits = 2)))
ggplot(plotTab, aes(x=condition, y=logVal, col = title)) +
geom_point() +
geom_line(aes(group=patID), linetype = "dotted") +
facet_wrap(~title) + ggtitle(nn) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "none")
})
cowplot::plot_grid(plotlist = pList, ncol=2)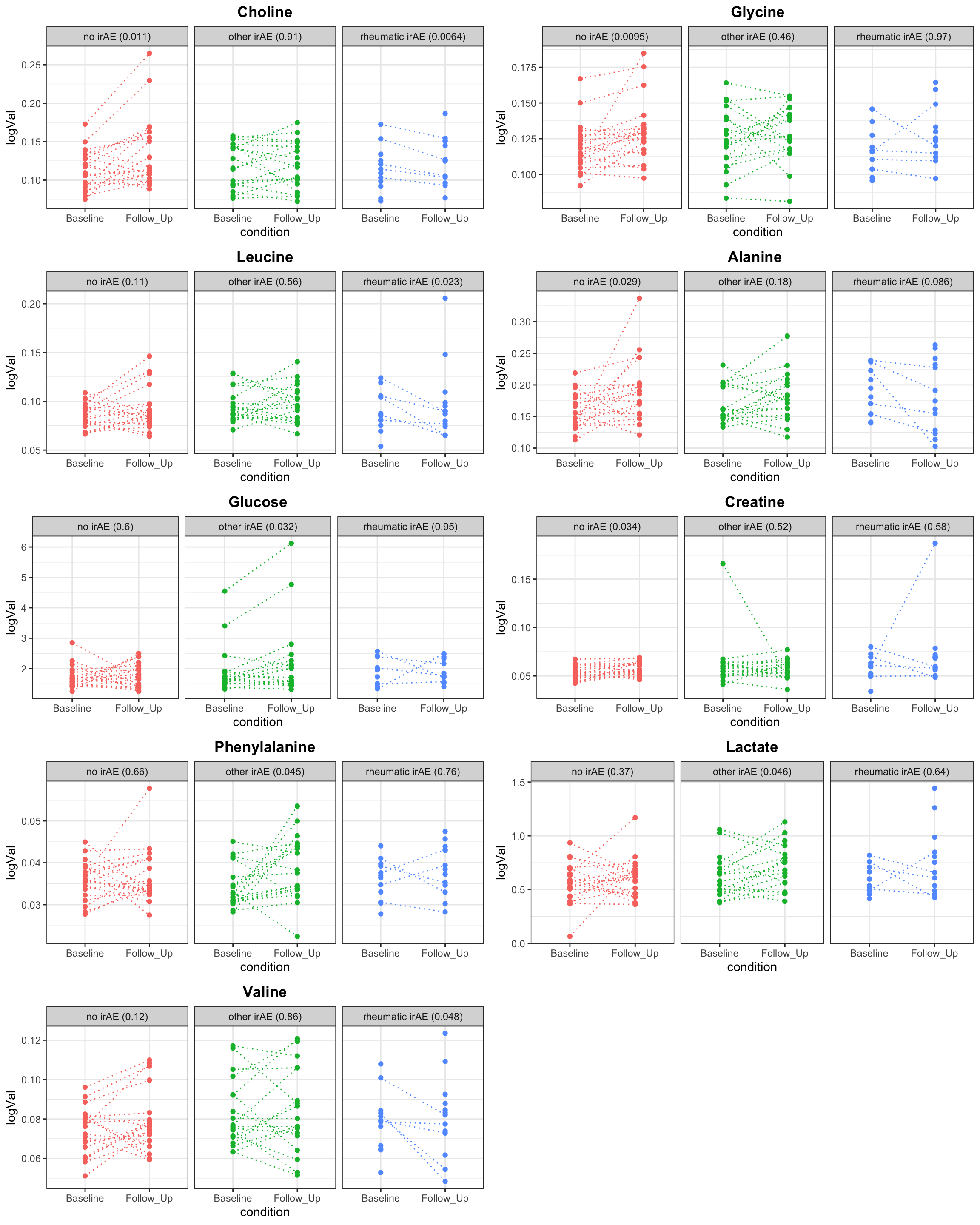
Healthy controls vs baseline of all cancer groups
CBA data
Remove one batch to avoid batch effect
testTab <- filter(fullTab, assay == "CBA", as.character(dateOfAcquisition_CBA) != "2022-06-08") %>%
mutate(logVal = glog2(value)) %>%
filter(Group !="rheumatic control", condition %in% c("Baseline","noMalignancy")) %>%
mutate(testGroup = ifelse(condition == "Baseline", "cancer", "healthy"))Test results
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~t.test(logVal ~ testGroup, var.equal=TRUE, data = .))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name, estimate, p.value) %>%
arrange(p.value)
resTab.sig <- filter(resTab, p.value <= 0.05)
resTab.sig# A tibble: 3 × 3
# Groups: name [3]
name estimate p.value
<chr> <dbl> <dbl>
1 sCD40L 1.85 0.00190
2 GM-CSF 0.988 0.0269
3 IFN-alpha2 0.645 0.0427 Plot significant associations (p<=0.05)
resTab.sig <- filter(resTab, p.value <=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=testGroup, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = Group)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "bottom") +
facet_wrap(~name, scales = "free")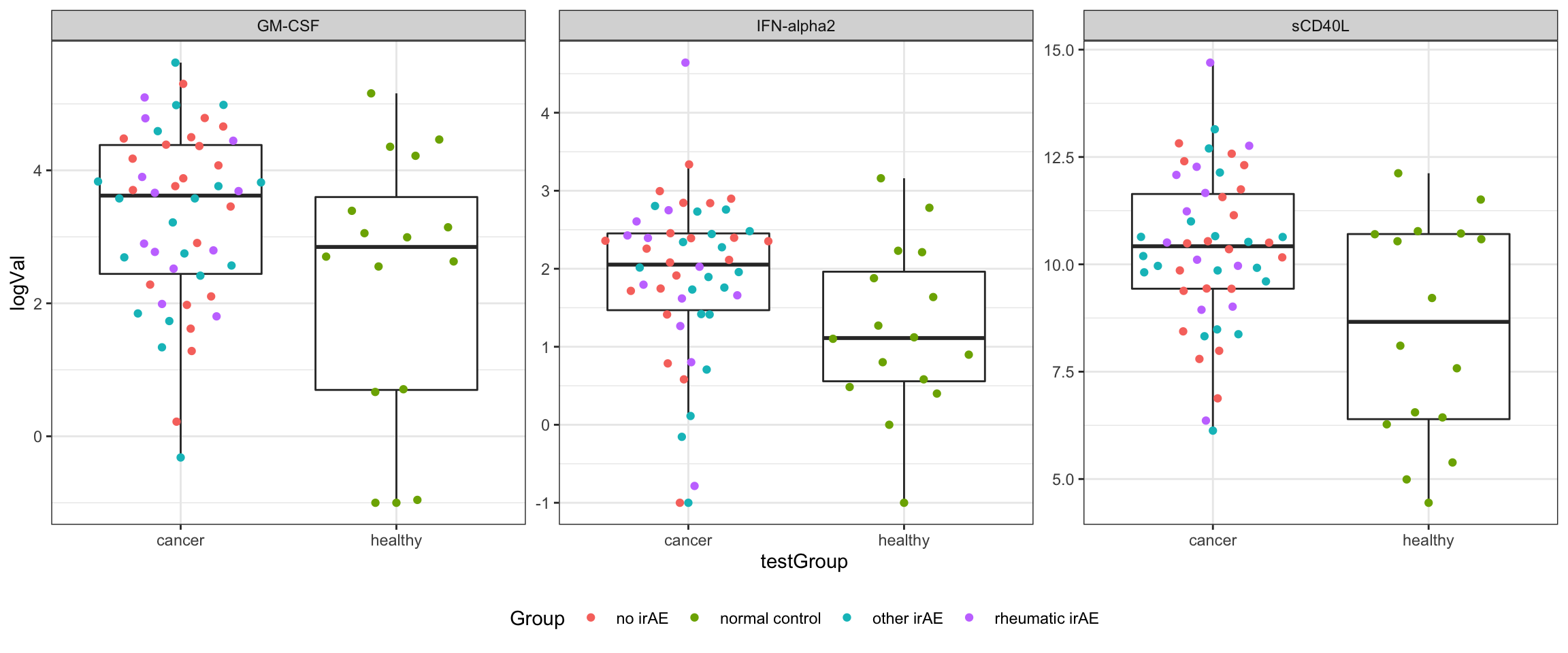
NMR data
testTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = value) %>%
filter(Group !="rheumatic control", condition %in% c("Baseline","noMalignancy")) %>%
mutate(testGroup = ifelse(condition == "Baseline", "cancer", "healthy"))Test results
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~t.test(logVal ~ testGroup, var.equal=TRUE, data = .))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name, estimate, p.value) %>%
arrange(p.value)
resTab.sig <- filter(resTab, p.value <= 0.01)
resTab.sig# A tibble: 23 × 3
# Groups: name [23]
name estimate p.value
<chr> <dbl> <dbl>
1 Glutamine 0.0728 2.07e-23
2 Histidine -0.0167 2.23e-21
3 L6L1 0.126 3.54e-20
4 Succinate -0.00474 1.32e-17
5 Choline 0.0478 2.15e-14
6 Aspartate 0.0319 2.06e-13
7 L3L1 -0.0765 3.36e-13
8 L2L1 0.417 2.32e-12
9 Glutamate -0.0451 3.58e-12
10 Formate -0.00335 3.44e- 9
# … with 13 more rows
# ℹ Use `print(n = ...)` to see more rowsPlot significant associations (p<=0.01)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=testGroup, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = Group)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "bottom") +
facet_wrap(~name, scales = "free", ncol = 3)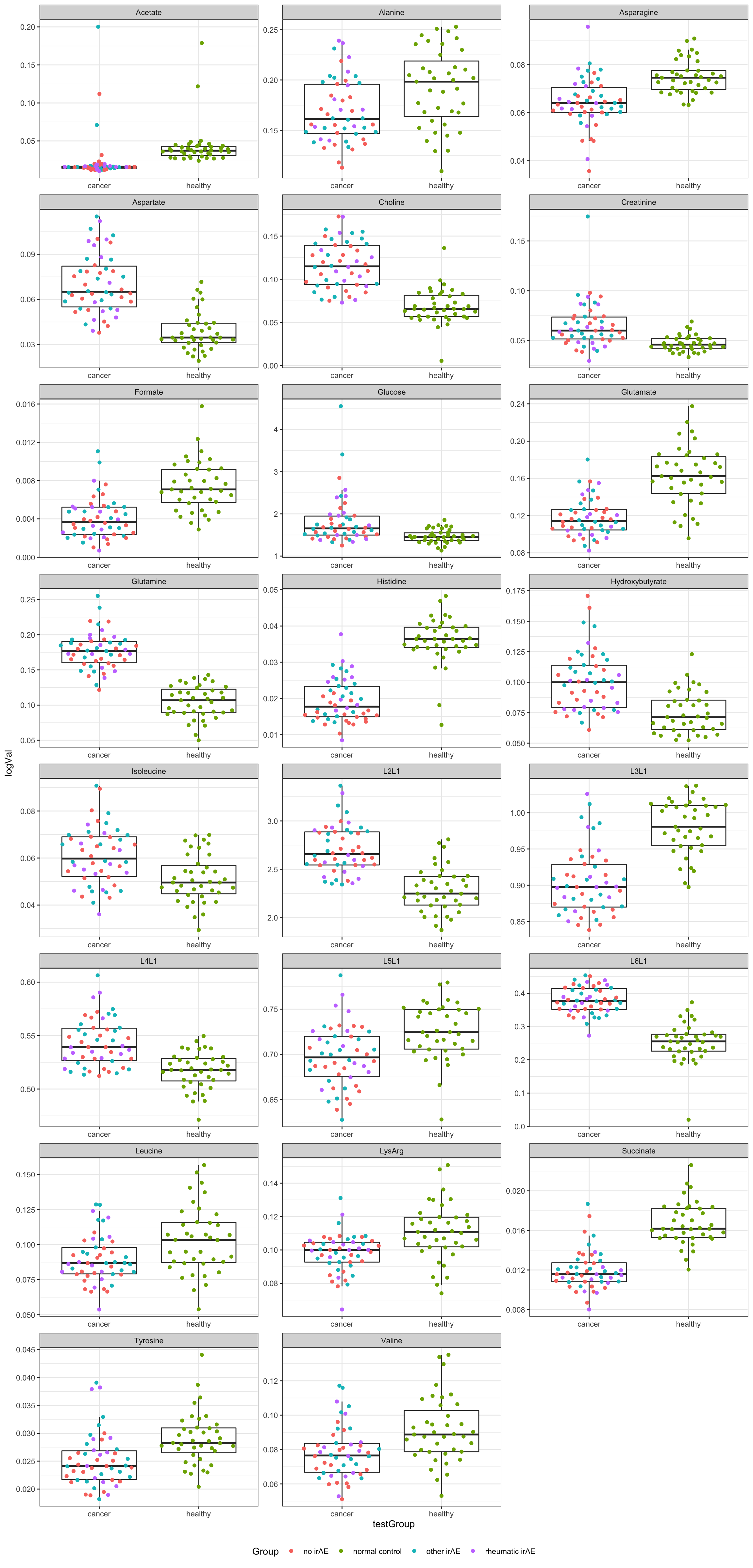
Rheuma irAE at follow-up vs Rheumatic non-cancer
CBA data
Remove one batch to avoid batch effect
testTab <- filter(fullTab, assay == "CBA", as.character(dateOfAcquisition_CBA) != "2022-06-08") %>%
mutate(logVal = glog2(value)) %>%
filter(Group %in% c("rheumatic control","rheumatic irAE"), condition %in% c("Follow_Up","noMalignancy"))resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~t.test(logVal ~ Group, var.equal=TRUE, data = .))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name, estimate, p.value) %>%
arrange(p.value)
resTab.sig <- filter(resTab, p.value <= 0.05)
resTab.sig# A tibble: 11 × 3
# Groups: name [11]
name estimate p.value
<chr> <dbl> <dbl>
1 IL-33 2.96 0.0000255
2 IL-12p70 1.72 0.000301
3 IL-23 2.09 0.00161
4 IL-10 2.28 0.00165
5 Perforin -0.648 0.00248
6 IL-8 1.87 0.00736
7 TGF-b1 -1.32 0.00787
8 IL-17A 0.531 0.0160
9 IFN-gamma 0.795 0.0338
10 GM-CSF -1.06 0.0449
11 TNF-alpha 1.13 0.0492 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = Group)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "bottom") +
facet_wrap(~name, scales = "free")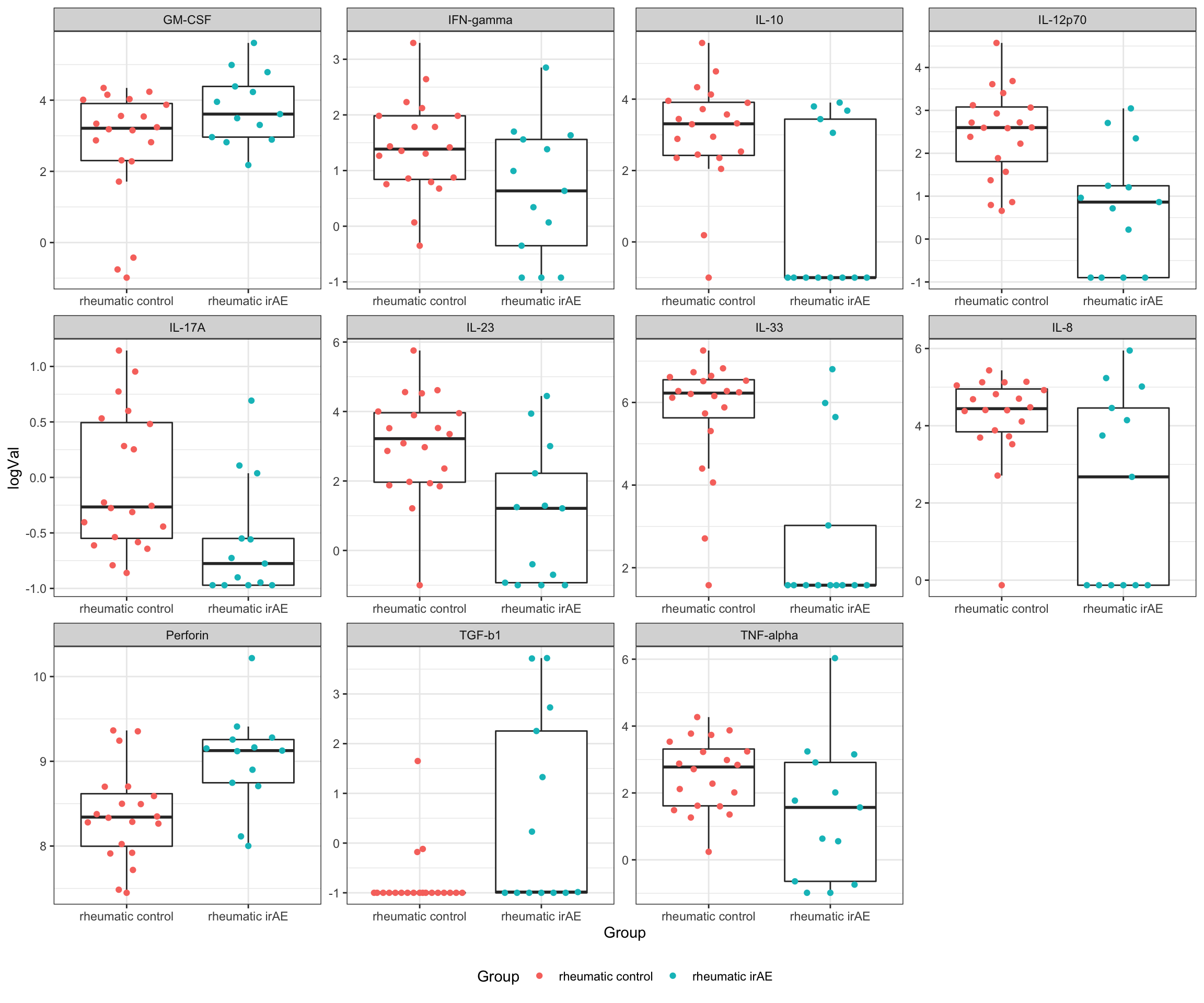
NMR data
Remove one batch to avoid batch effect
testTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = glog2(value)) %>%
filter(Group %in% c("rheumatic control","rheumatic irAE"), condition %in% c("Follow_Up","noMalignancy"))resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~t.test(logVal ~ Group, var.equal=TRUE, data = .))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% select(name, estimate, p.value) %>%
arrange(p.value)
resTab.sig <- filter(resTab, p.value <= 0.05)
resTab.sig# A tibble: 15 × 3
# Groups: name [15]
name estimate p.value
<chr> <dbl> <dbl>
1 Glutamine -0.0862 0.00000102
2 Choline -0.0673 0.0000236
3 Acetate 0.0164 0.000213
4 Formate 0.00425 0.000266
5 Creatinine -0.0441 0.000782
6 L3L1 0.0717 0.00178
7 Glucose -0.313 0.00250
8 L6L1 -0.118 0.00383
9 Glutamate 0.0509 0.00406
10 Glycerol -0.0172 0.00436
11 Threonine -0.0308 0.00439
12 Aspartate -0.0340 0.0242
13 Tyrosine 0.00579 0.0294
14 Creatine -0.0266 0.0294
15 Succinate 0.00371 0.0454 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=Group, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = Group)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "bottom") +
facet_wrap(~name, scales = "free")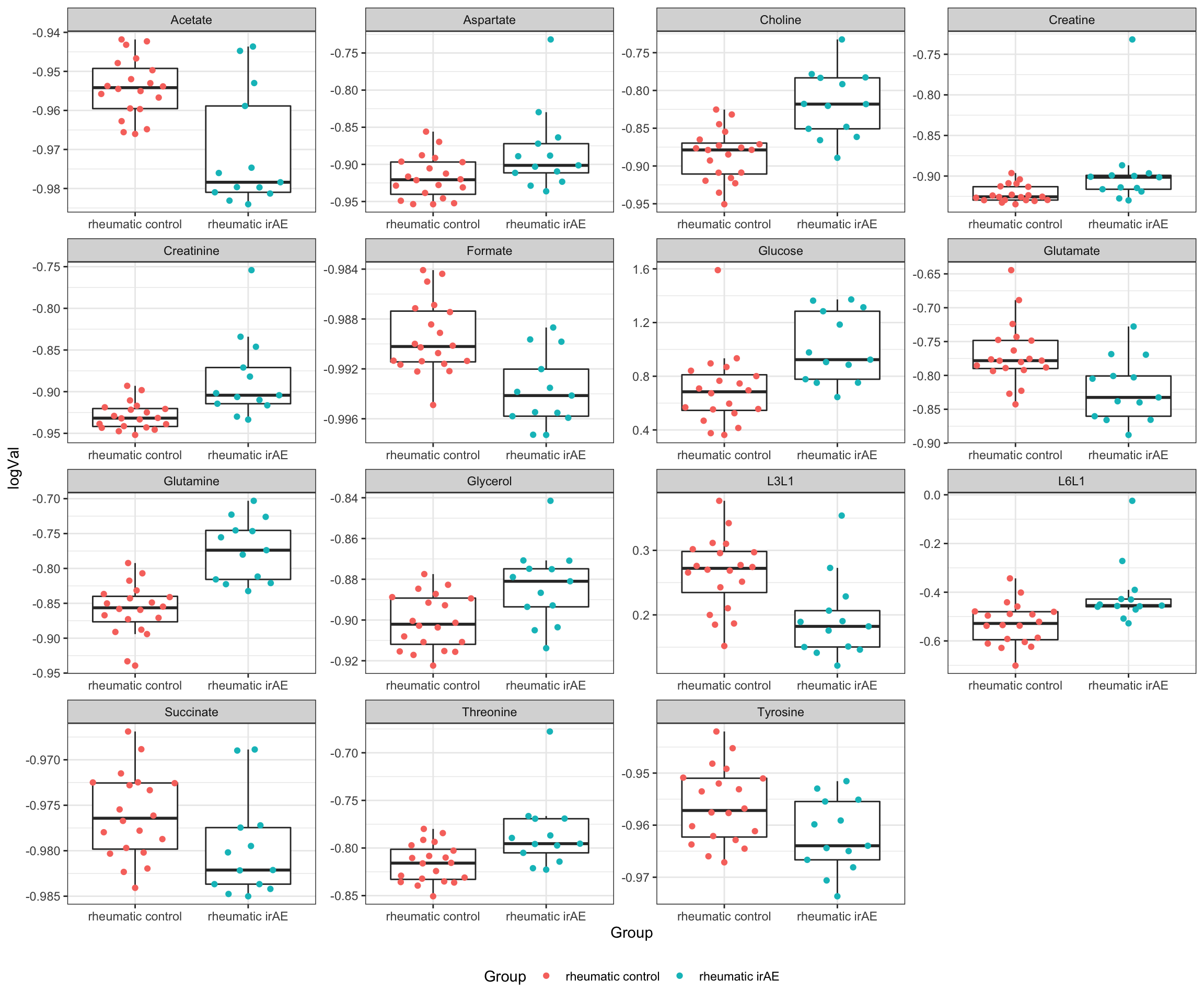
Test for interaction between Cancer and Rheumatic irAE (at follow-up)
CBA data
Remove one batch to avoid batch effect
testTab <- filter(fullTab, assay == "CBA", as.character(dateOfAcquisition_CBA) != "2022-06-08") %>%
mutate(logVal = glog2(value)) %>%
filter(Group %in% c("rheumatic control","rheumatic irAE", "normal control", "no irAE"), condition %in% c("Follow_Up","noMalignancy")) %>%
mutate(cancer = ifelse(condition == "noMalignancy", "noCancer", "withCancer"),
irAE = ifelse(str_detect(Group, "rheumatic"),"Rheumatic", "noRheumatic")) %>%
mutate(category = paste0(cancer,"_",irAE))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~car::Anova(lm(logVal ~cancer+irAE+cancer:irAE,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res)Iteractions between cancer and rheumatic irAE
The below results show the significant interaction between cancer and rheumatic irAE on the molecular profile. A “significant interaction” means the whether the patients have cancer or irAE have non-additive effect, e.g one molecular is up-regulated in patients with irAE in cancer group but down-regulated (or unchanged) in non-cancer group.
resTab.inter <- resTab %>%
filter(term == "cancer:irAE") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05)
resTab.inter# A tibble: 9 × 2
# Groups: name [9]
name p.value
<chr> <dbl>
1 IL-33 0.0000392
2 IL-12p70 0.000399
3 IL-10 0.00175
4 Perforin 0.00587
5 IL-17A 0.0134
6 TGF-b1 0.0286
7 IL-23 0.0340
8 IFN-gamma 0.0389
9 IFN-alpha2 0.0482 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.inter$name)
ggplot(plotTab, aes(x=category, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = category)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name, scales = "free")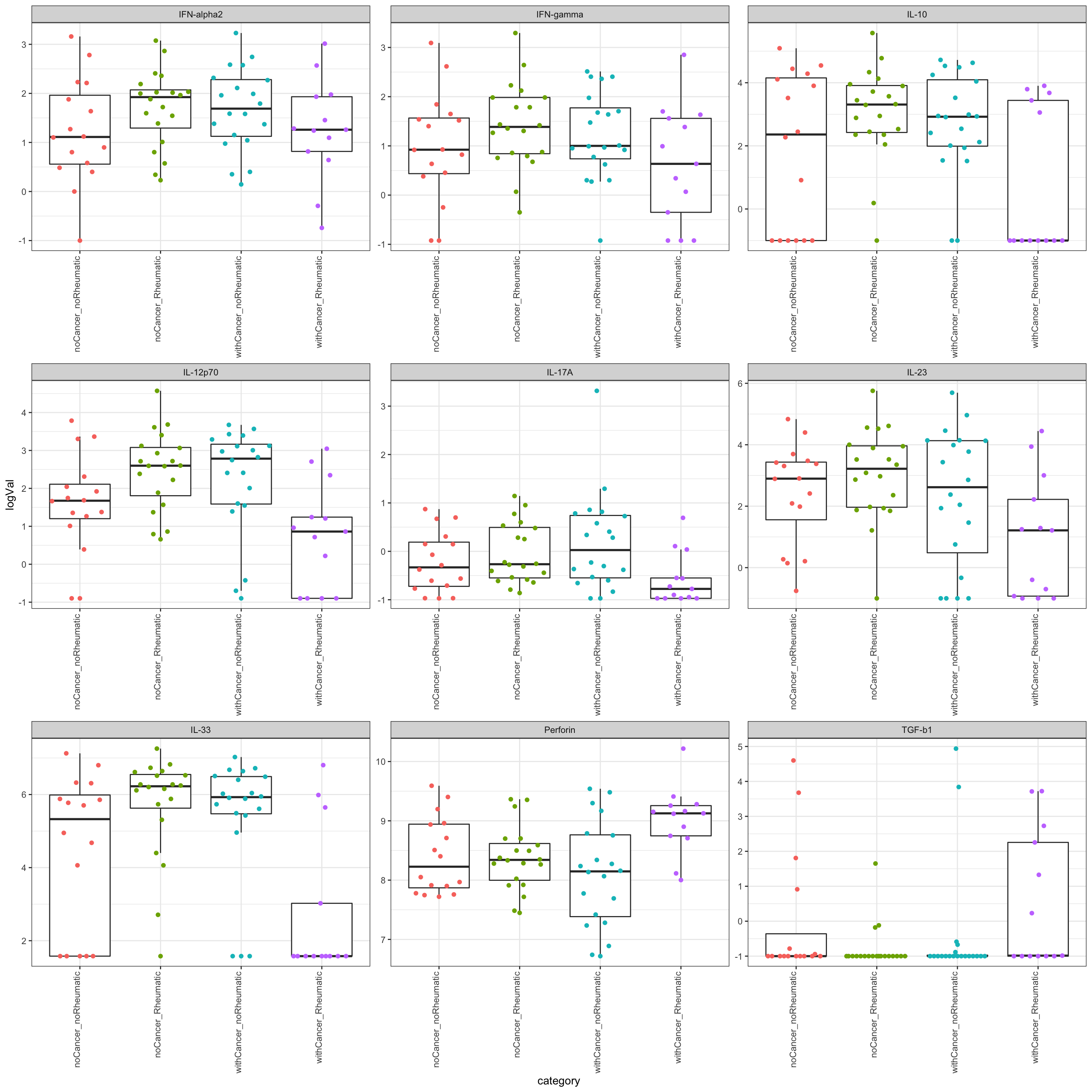
Different between cancer and non-cancer, regardless of irAE status
The below results show the molecules that show similar trend between cancer and non-cancer group, regardless of whether the patients have irAE or not.
resTab.sig <- resTab %>%
filter(term == "cancer") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05, !name %in% resTab.inter$name)
resTab.sig# A tibble: 6 × 2
# Groups: name [6]
name p.value
<chr> <dbl>
1 Granzyme 0.00220
2 sCD40L 0.0177
3 IL-8 0.0291
4 IL-18 0.0421
5 sFAS 0.0470
6 GM-CSF 0.0478 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=cancer, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = cancer)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name+irAE, scales = "free",ncol=4)
Different between irAE and no irAE, regardless of cancer
The below results show the molecules that show similar trend between rheumatic irAE and non-irAE group, regardless of whether the patients have cancer or not.
resTab.sig <- resTab %>%
filter(term == "irAE") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05, !name %in% resTab.inter$name)
resTab.sig# A tibble: 1 × 2
# Groups: name [1]
name p.value
<chr> <dbl>
1 sCD40L 0.00623Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=irAE, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = irAE)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name+cancer, scales = "free",ncol=4)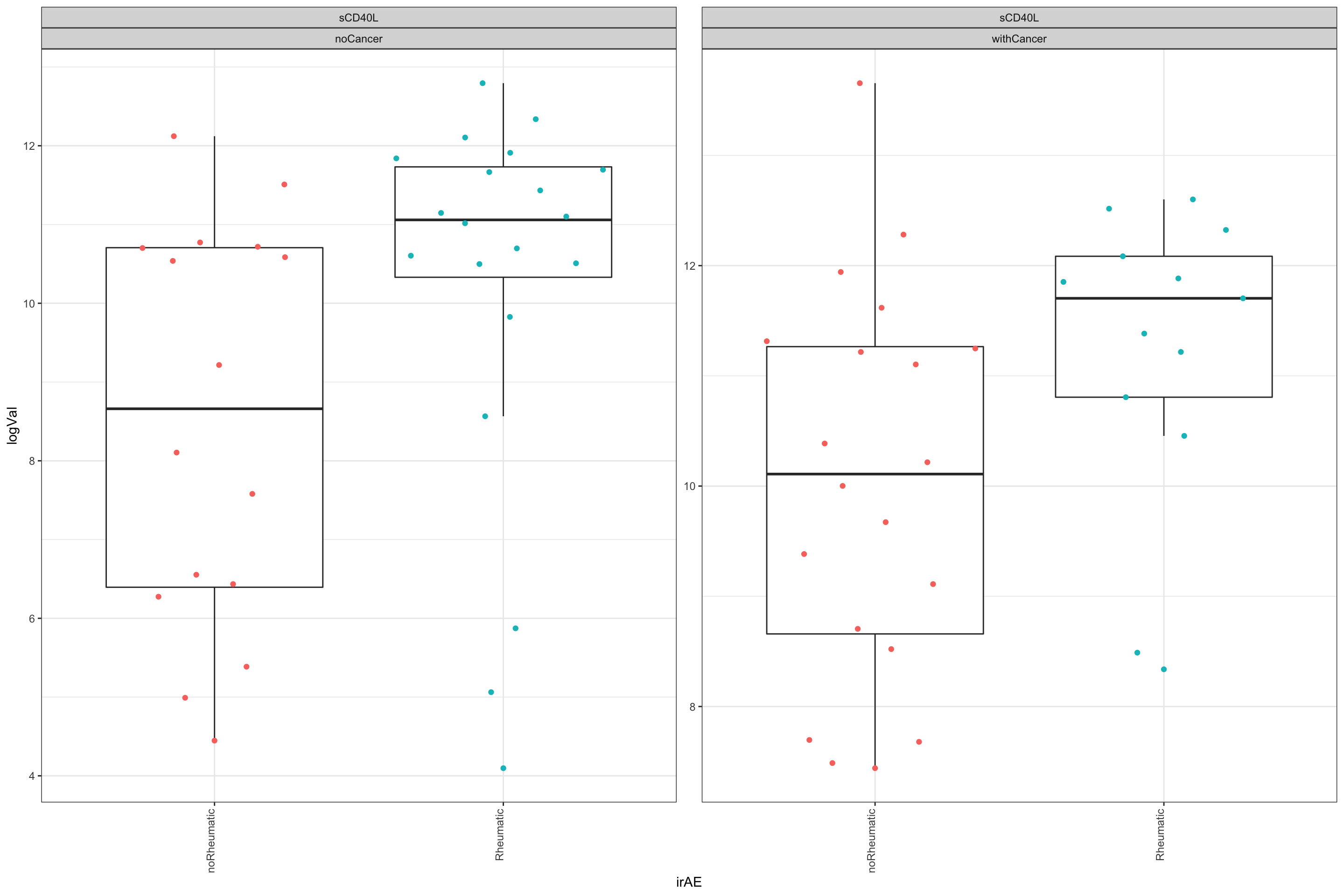
NMR data
The same analyses as for the CBA data.
testTab <- filter(fullTab, assay == "NMR") %>%
mutate(logVal = glog2(value)) %>%
filter(Group %in% c("rheumatic control","rheumatic irAE", "normal control", "no irAE"), condition %in% c("Follow_Up","noMalignancy")) %>%
mutate(cancer = ifelse(condition == "noMalignancy", "noCancer", "withCancer"),
irAE = ifelse(str_detect(Group, "rheumatic"),"Rheumatic", "noRheumatic")) %>%
mutate(category = paste0(cancer,"_",irAE))
resTab <- group_by(testTab, name) %>% nest() %>%
mutate(m = map(data, ~car::Anova(lm(logVal ~cancer+irAE+cancer:irAE,.)))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res)Iteractions between cancer and rheumatic irAE
resTab.inter <- resTab %>%
filter(term == "cancer:irAE") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05)
resTab.inter# A tibble: 6 × 2
# Groups: name [6]
name p.value
<chr> <dbl>
1 Histidine 0.0000000209
2 Asparagine 0.000616
3 Creatine 0.000923
4 L2L1 0.0132
5 L5L1 0.0359
6 Glutamine 0.0495 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.inter$name)
ggplot(plotTab, aes(x=category, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = category)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name, scales = "free")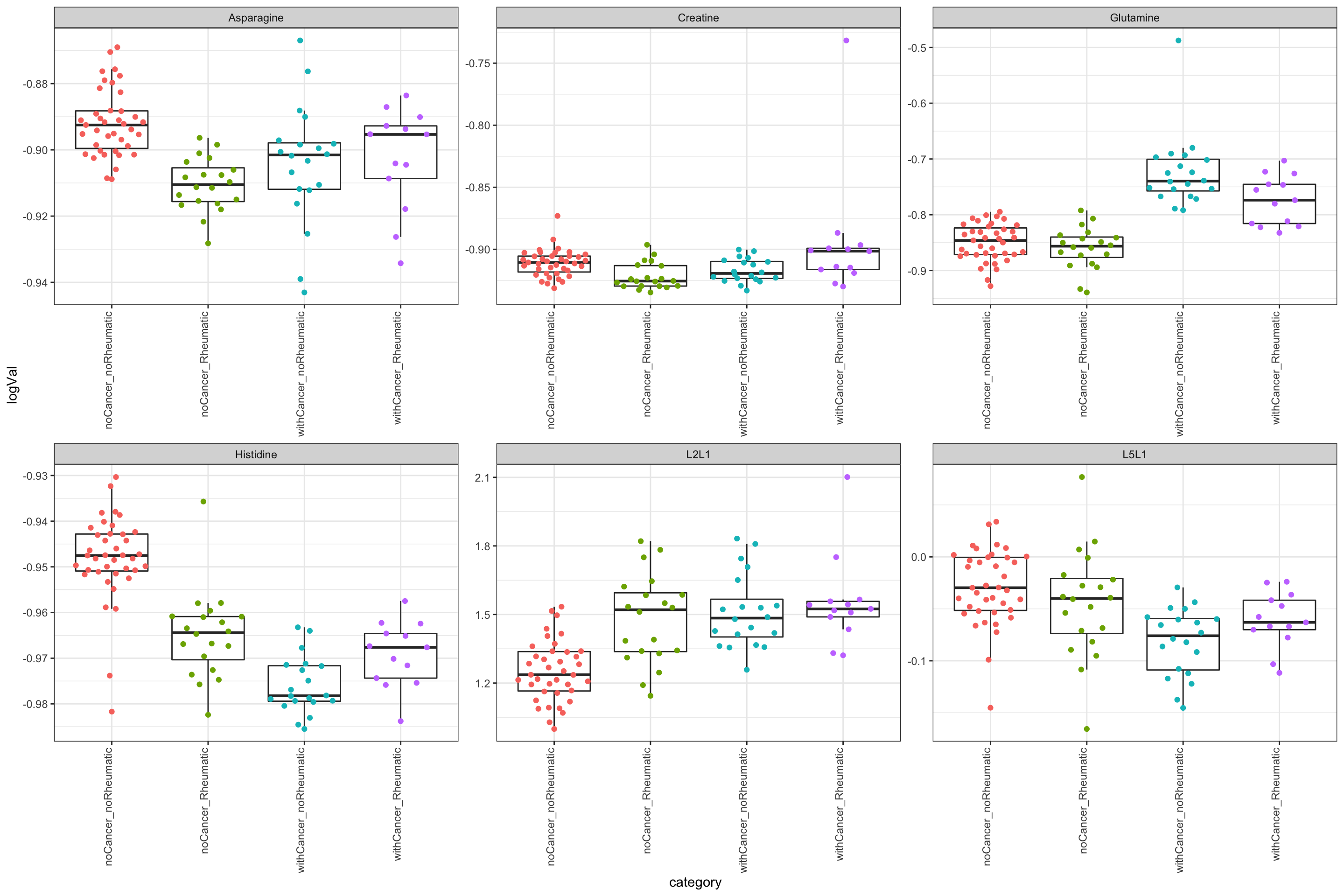
Different between cancer and non-cancer, regardless of irAE status
resTab.sig <- resTab %>%
filter(term == "cancer") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05, !name %in% resTab.inter$name)
resTab.sig# A tibble: 18 × 2
# Groups: name [18]
name p.value
<chr> <dbl>
1 Choline 9.26e-15
2 L6L1 3.05e-13
3 L3L1 6.89e-12
4 Formate 2.10e-10
5 Creatinine 1.62e- 9
6 Aspartate 9.59e- 9
7 Succinate 2.07e- 7
8 Glucose 6.79e- 7
9 Acetate 7.61e- 7
10 Glutamate 2.39e- 6
11 Hydroxybutyrate 2.59e- 5
12 L4L1 4.09e- 5
13 Isoleucine 8.67e- 5
14 Threonine 5.47e- 4
15 Tyrosine 3.63e- 3
16 Valine 4.44e- 3
17 Glycine 8.65e- 3
18 LysArg 2.06e- 2Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=cancer, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = cancer)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name+irAE, scales = "free", ncol=4)
Different between irAE and no irAE, regardless of cancer
resTab.sig <- resTab %>%
filter(term == "irAE") %>%
select(name, p.value) %>%
arrange(p.value) %>%
filter(p.value <= 0.05, !name %in% resTab.inter$name)
resTab.sig# A tibble: 4 × 2
# Groups: name [4]
name p.value
<chr> <dbl>
1 L6L1 0.0000419
2 Lactate 0.000267
3 Aspartate 0.00431
4 L4L1 0.00614 Plot significant associations (p<=0.05)
plotTab <- filter(testTab, name %in% resTab.sig$name)
ggplot(plotTab, aes(x=irAE, y=logVal)) +
geom_boxplot(outlier.shape = NA) +
ggbeeswarm::geom_quasirandom(aes(col = irAE)) +
theme_bw() +
theme(plot.title = element_text(face="bold", hjust = 0.5),
legend.position = "no",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0)) +
facet_wrap(~name+cancer, scales = "free", ncol=4)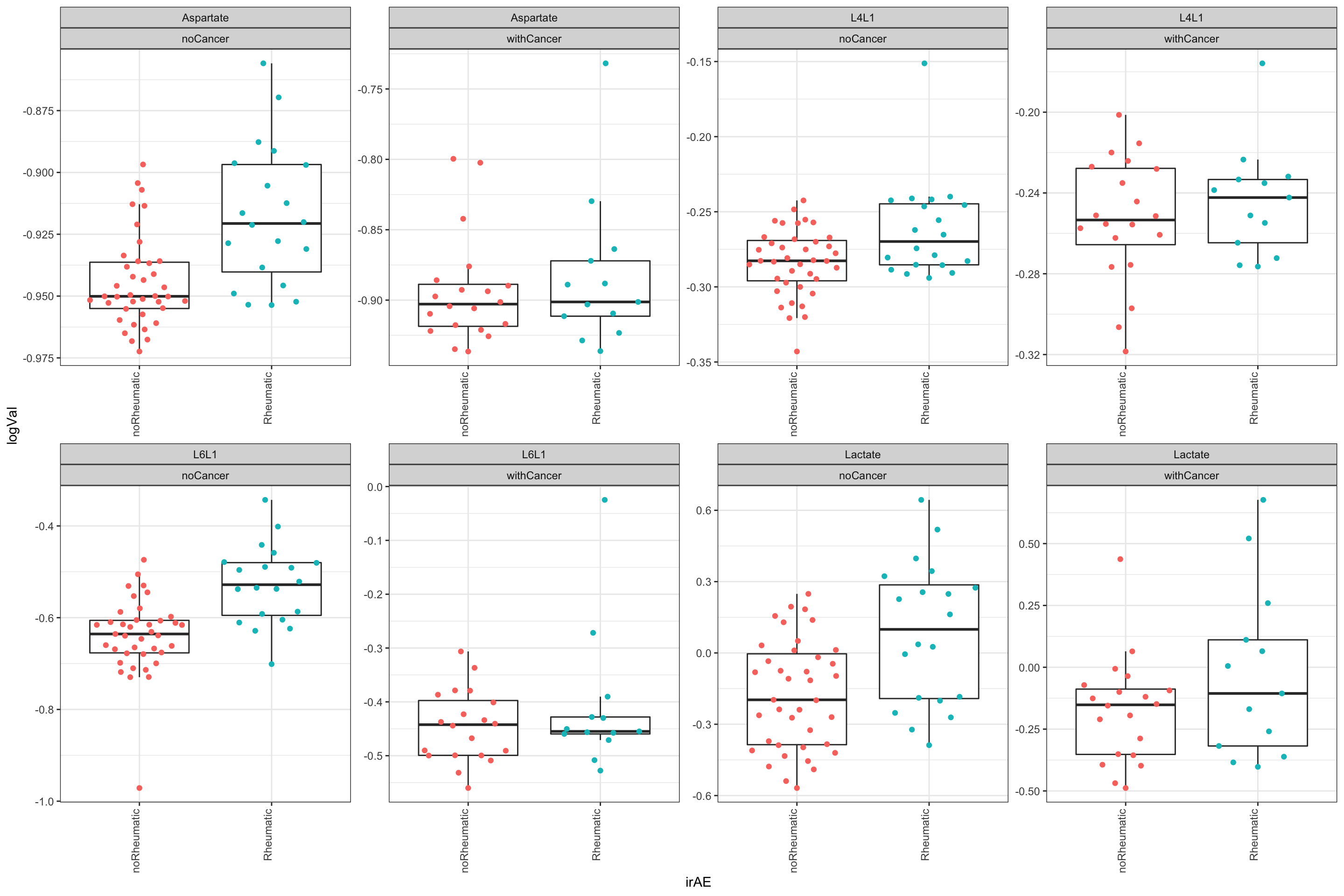
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.0
[3] dplyr_1.0.9 purrr_0.3.4
[5] readr_2.1.2 tidyr_1.2.0
[7] tibble_3.1.8 ggplot2_3.3.6
[9] tidyverse_1.3.2 jyluMisc_0.1.5
[11] vsn_3.64.0 pheatmap_1.0.12
[13] MultiAssayExperiment_1.22.0 SummarizedExperiment_1.26.1
[15] Biobase_2.56.0 GenomicRanges_1.48.0
[17] GenomeInfoDb_1.32.2 IRanges_2.30.0
[19] S4Vectors_0.34.0 BiocGenerics_0.42.0
[21] MatrixGenerics_1.8.1 matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] readxl_1.4.0 backports_1.4.1 fastmatch_1.1-3
[4] drc_3.0-1 workflowr_1.7.0 igraph_1.3.4
[7] shinydashboard_0.7.2 splines_4.2.0 BiocParallel_1.30.3
[10] TH.data_1.1-1 digest_0.6.29 htmltools_0.5.3
[13] fansi_1.0.3 magrittr_2.0.3 googlesheets4_1.0.0
[16] cluster_2.1.3 tzdb_0.3.0 limma_3.52.2
[19] modelr_0.1.8 sandwich_3.0-2 piano_2.12.0
[22] colorspace_2.0-3 rvest_1.0.2 haven_2.5.0
[25] xfun_0.31 crayon_1.5.1 RCurl_1.98-1.7
[28] jsonlite_1.8.0 survival_3.4-0 zoo_1.8-10
[31] glue_1.6.2 survminer_0.4.9 gtable_0.3.0
[34] gargle_1.2.0 zlibbioc_1.42.0 XVector_0.36.0
[37] DelayedArray_0.22.0 car_3.1-0 abind_1.4-5
[40] scales_1.2.0 mvtnorm_1.1-3 DBI_1.1.3
[43] relations_0.6-12 rstatix_0.7.0 Rcpp_1.0.9
[46] plotrix_3.8-2 xtable_1.8-4 preprocessCore_1.58.0
[49] km.ci_0.5-6 DT_0.23 httr_1.4.3
[52] htmlwidgets_1.5.4 fgsea_1.22.0 gplots_3.1.3
[55] RColorBrewer_1.1-3 ellipsis_0.3.2 farver_2.1.1
[58] pkgconfig_2.0.3 sass_0.4.2 dbplyr_2.2.1
[61] utf8_1.2.2 labeling_0.4.2 tidyselect_1.1.2
[64] rlang_1.0.4 later_1.3.0 munsell_0.5.0
[67] cellranger_1.1.0 tools_4.2.0 visNetwork_2.1.0
[70] cachem_1.0.6 cli_3.3.0 generics_0.1.3
[73] broom_1.0.0 evaluate_0.15 fastmap_1.1.0
[76] yaml_2.3.5 knitr_1.39 fs_1.5.2
[79] survMisc_0.5.6 caTools_1.18.2 mime_0.12
[82] slam_0.1-50 xml2_1.3.3 compiler_4.2.0
[85] rstudioapi_0.13 beeswarm_0.4.0 affyio_1.66.0
[88] ggsignif_0.6.3 marray_1.74.0 reprex_2.0.1
[91] bslib_0.4.0 stringi_1.7.8 highr_0.9
[94] lattice_0.20-45 Matrix_1.4-1 shinyjs_2.1.0
[97] KMsurv_0.1-5 vctrs_0.4.1 pillar_1.8.0
[100] lifecycle_1.0.1 BiocManager_1.30.18 jquerylib_0.1.4
[103] data.table_1.14.2 cowplot_1.1.1 bitops_1.0-7
[106] httpuv_1.6.5 R6_2.5.1 affy_1.74.0
[109] promises_1.2.0.1 KernSmooth_2.23-20 gridExtra_2.3
[112] vipor_0.4.5 codetools_0.2-18 MASS_7.3-58
[115] gtools_3.9.3 exactRankTests_0.8-35 assertthat_0.2.1
[118] rprojroot_2.0.3 withr_2.5.0 multcomp_1.4-19
[121] GenomeInfoDbData_1.2.8 parallel_4.2.0 hms_1.1.1
[124] grid_4.2.0 rmarkdown_2.14 carData_3.0-5
[127] googledrive_2.0.0 git2r_0.30.1 maxstat_0.7-25
[130] ggpubr_0.4.0 sets_1.0-21 lubridate_1.8.0
[133] shiny_1.7.2 ggbeeswarm_0.6.0