Investigate the difference between healthy samples and samples with injury (at baseline)
Junyan Lu
17 May 2024
Last updated: 2024-05-17
Checks: 5 1
Knit directory:
SpinalCord_proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Data preparation
Regress-out unwanted variations using SVA method
seProt_corr <- seProt
patAnno <- colData(seProt_corr)
patAnno$Visit <- factor(ifelse(is.na(patAnno$Visit),0, patAnno$Visit))
patAnno$delta_UEMS <- ifelse(is.na(patAnno$delta_UEMS),0, patAnno$delta_UEMS)
patAnno$UEMS <- ifelse(is.na(patAnno$UEMS),0, patAnno$UEMS)
patAnno$AIS <- ifelse(is.na(patAnno$AIS),"None", patAnno$AIS)
patAnno$treatVis <- paste0(patAnno$Treatment, patAnno$Visit)
patAnno$nodeGroup <- factor(patAnno$nodeGroup)
mod <- model.matrix(~ treatVis + AIS + UEMS + delta_UEMS, patAnno)
exprMat <- assays(seProt_corr)[[2]]
svaObj <- sva::sva(exprMat, mod)Number of significant surrogate variables is: 10
Iteration (out of 5 ):1 2 3 4 5 assays(seProt_corr)[[1]] <- limma::removeBatchEffect(assay(seProt_corr), covariates = svaObj$sv)
assays(seProt_corr)[[2]] <- limma::removeBatchEffect(assays(seProt_corr)[[2]], covariates = svaObj$sv)Subset for baseline samples
#protSub <- seProt_corr[,seProt_corr$Visit == 3 | is.na(seProt_corr$Visit)]
protSub <- prepareProt(seProt_corr, filterCondi = list(Visit = c(3,NA)), perNA = 0.5)[1] "Number of proteins: 377, number of samples: 131"protSub$group <- ifelse(is.na(protSub$Visit),"control","injury")
protSub$libSize <- colSums(assay(protSub),na.rm=TRUE)PCA
exprMat <- assays(protSub)[["imputed"]]
smpAnno <- colData(protSub) %>% as_tibble()
pcRes <- prcomp(t(exprMat), scale. = FALSE, center = TRUE)
pcTab <- pcRes$x[,1:20] %>%
as_tibble(rownames = "sampleID")
plotTab <- pcTab %>%
left_join(smpAnno)
varExp <- pcRes$sdev^2/sum(pcRes$sdev^2) * 100Test associations between PCA and metadata
metaTab <- smpAnno %>%
select(sampleID, group, UEMS, SEX, AGE, AIS, libSize, delta_UEMS, nodeGroup)
resTab <- jyluMisc::testAssociation(pcTab, metaTab, joinID = "sampleID") %>%
filter(p<0.05)
head(resTab) var1 var2 p p.adj
1 PC1 group 1.299342e-57 2.078946e-55
2 PC1 nodeGroup 8.006588e-57 6.405270e-55
3 PC2 AIS 1.044130e-04 5.568695e-03
4 PC2 UEMS 4.873588e-04 1.949435e-02
5 PC9 libSize 1.054214e-03 3.373486e-02
6 PC2 nodeGroup 2.043374e-03 5.448997e-02Plots
PC1 versus PC2
ggplot(plotTab, aes(x=PC1, y=PC2, color = group, shape = group)) +
geom_point(size=2) +
xlab(sprintf("PC1 (%1.2f%%)",varExp[1])) +
ylab(sprintf("PC2 (%1.2f%%)",varExp[2])) +
theme_full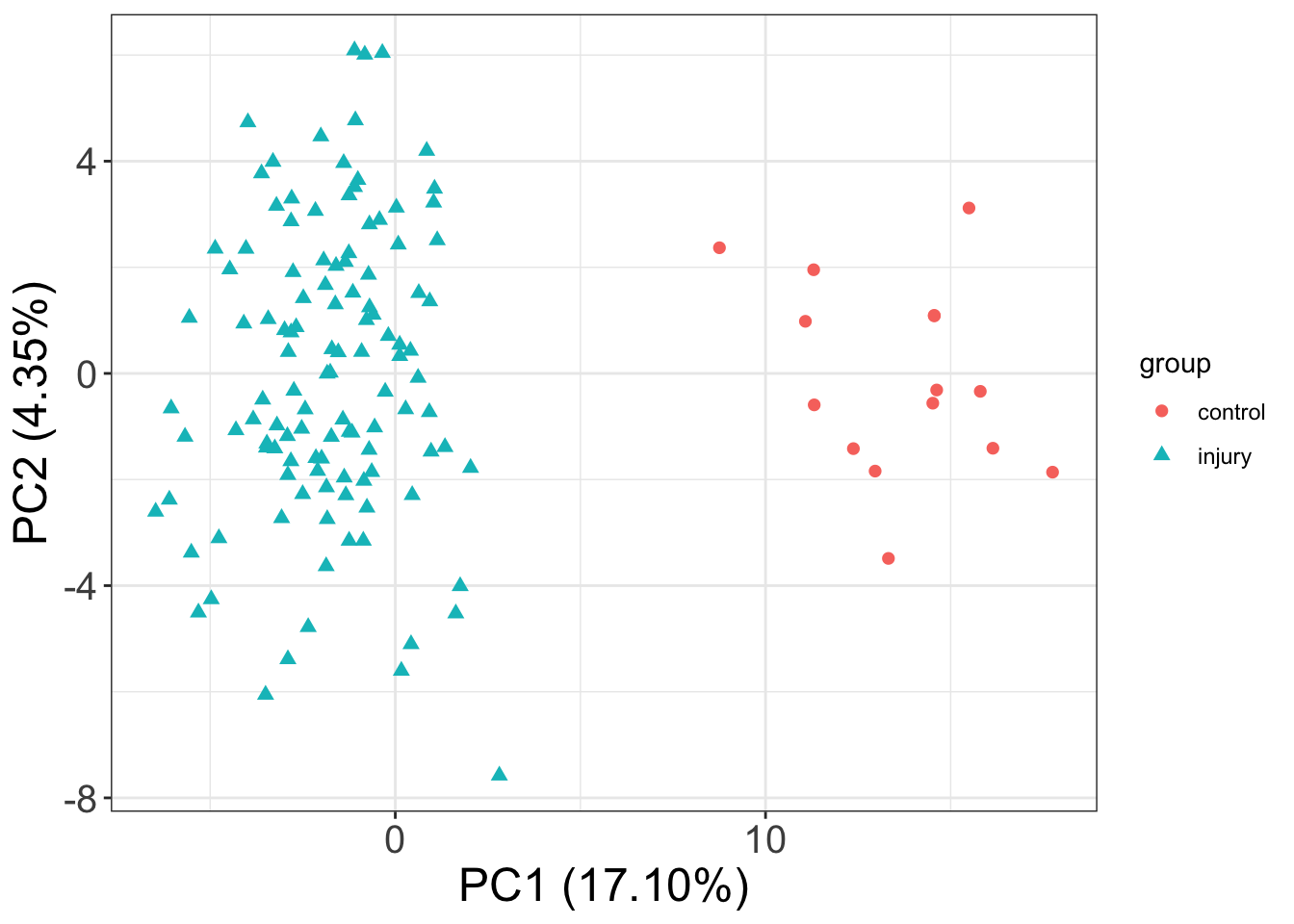 Control and injury samples can be clearly separated
Control and injury samples can be clearly separated
Differentially expressed proteins between control and injury samples
P-Value histogram
designMat <- model.matrix(~group, colData(protSub))
resTab <- testDiff(protSub, design = designMat, assayName = "imputed",
coef = "groupinjury", method = "limma")
hist(resTab$pval, main = "P-Value histogram")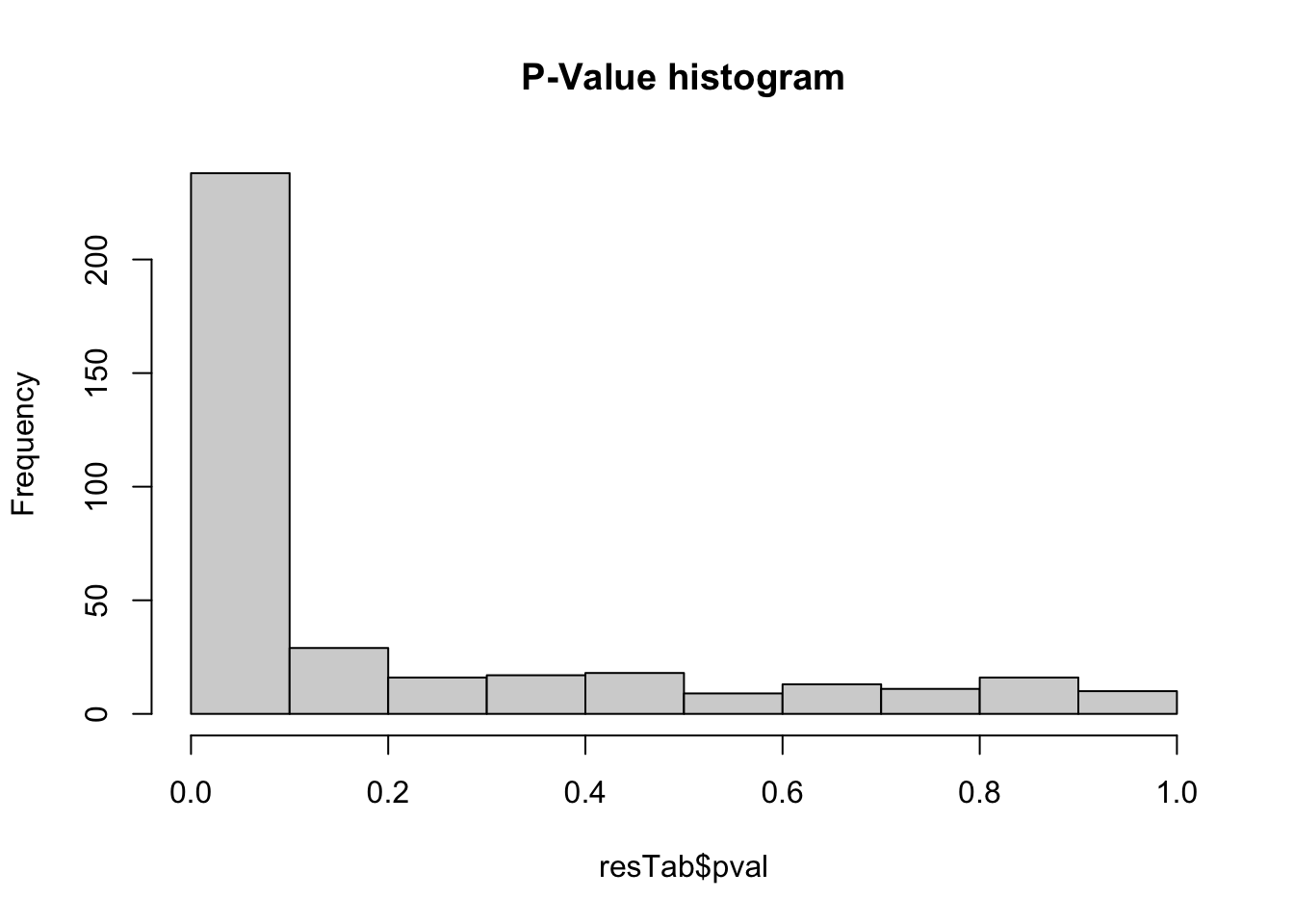
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
List of significant associations
5% FDR as cut-off
filter(resTab, adj_pval <= 0.05) %>% mutate(across(where(is.numeric), formatC, digits=2)) %>%
select(name, symbol, pval, adj_pval, diff, n_obs) %>%
DT::datatable()Boxplot of top associations
pList <- lapply(seq(9), function(i) {
rec <- resTab[i,]
plotTab <- tibble(expr = assays(protSub)[["imputed"]][rec$name,],
group = protSub$group)
ggplot(plotTab, aes(x=group, y = expr)) +
geom_boxplot(aes(fill = group), alpha=0.5) +
ggbeeswarm::geom_beeswarm() +
ggtitle(sprintf("%s (P=%s)",rec$symbol, formatC(rec$pval, digits = 2))) +
theme_full +
theme(legend.position = "none") +
xlab("") + ylab("Expression")
})
cowplot::plot_grid(plotlist = pList, ncol=3)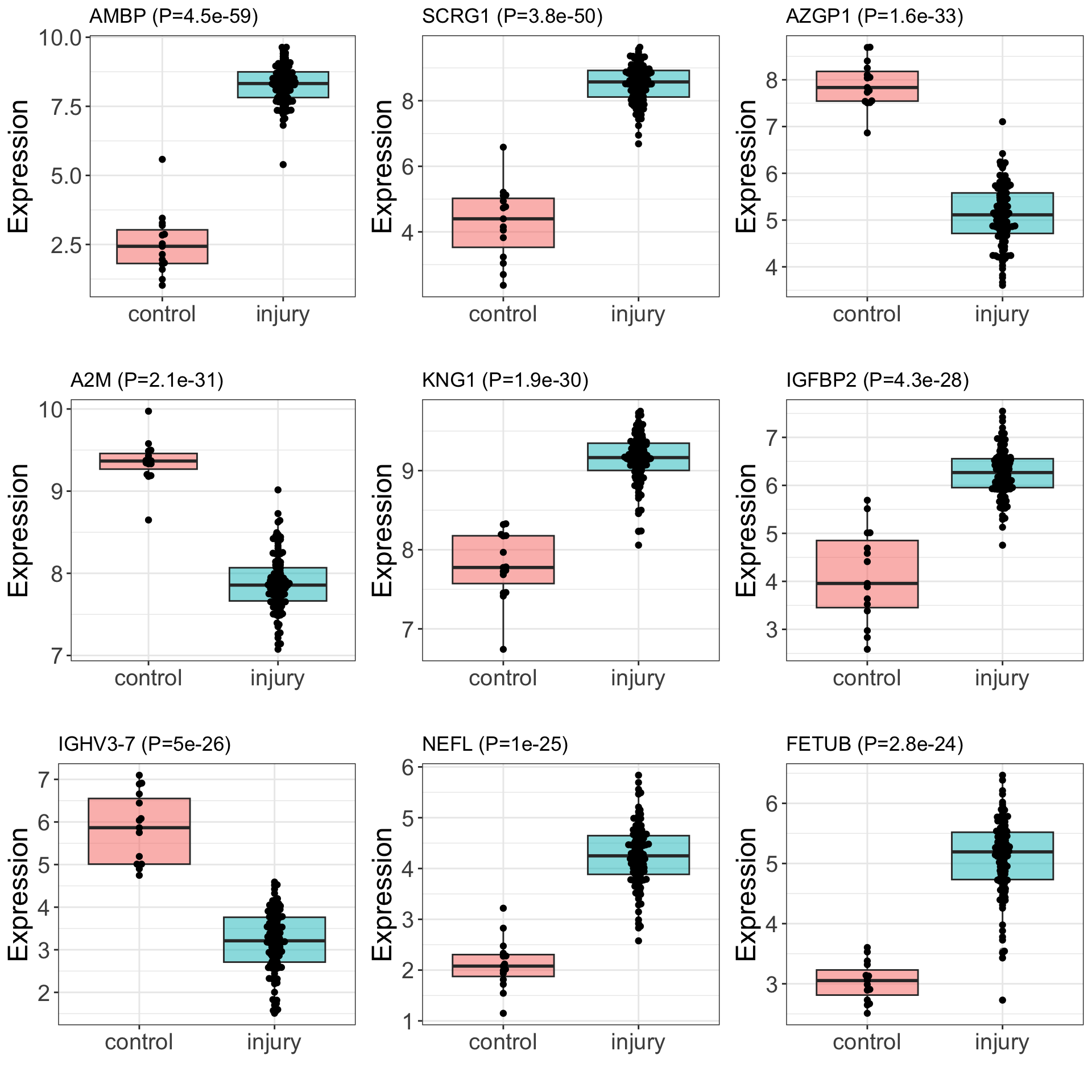
Enrichment analysis
All ranked genes are used. Pathway at 5% FDR level.
set.seed(2024)
gmts = list(GO_BiologicalProcess = "../data/gmts/c5.go.bp.v2023.2.Hs.symbols.gmt",
GO_MolecularFunction = "../data/gmts/c5.go.mf.v2023.2.Hs.symbols.gmt")
plotList <- runGeneSetEnrichment(resTab, gmts, genePCut = 0.1, pCutSet = 0.05, setFdr = FALSE, method = "gsea", collapsePathway = FALSE)
plotList$plot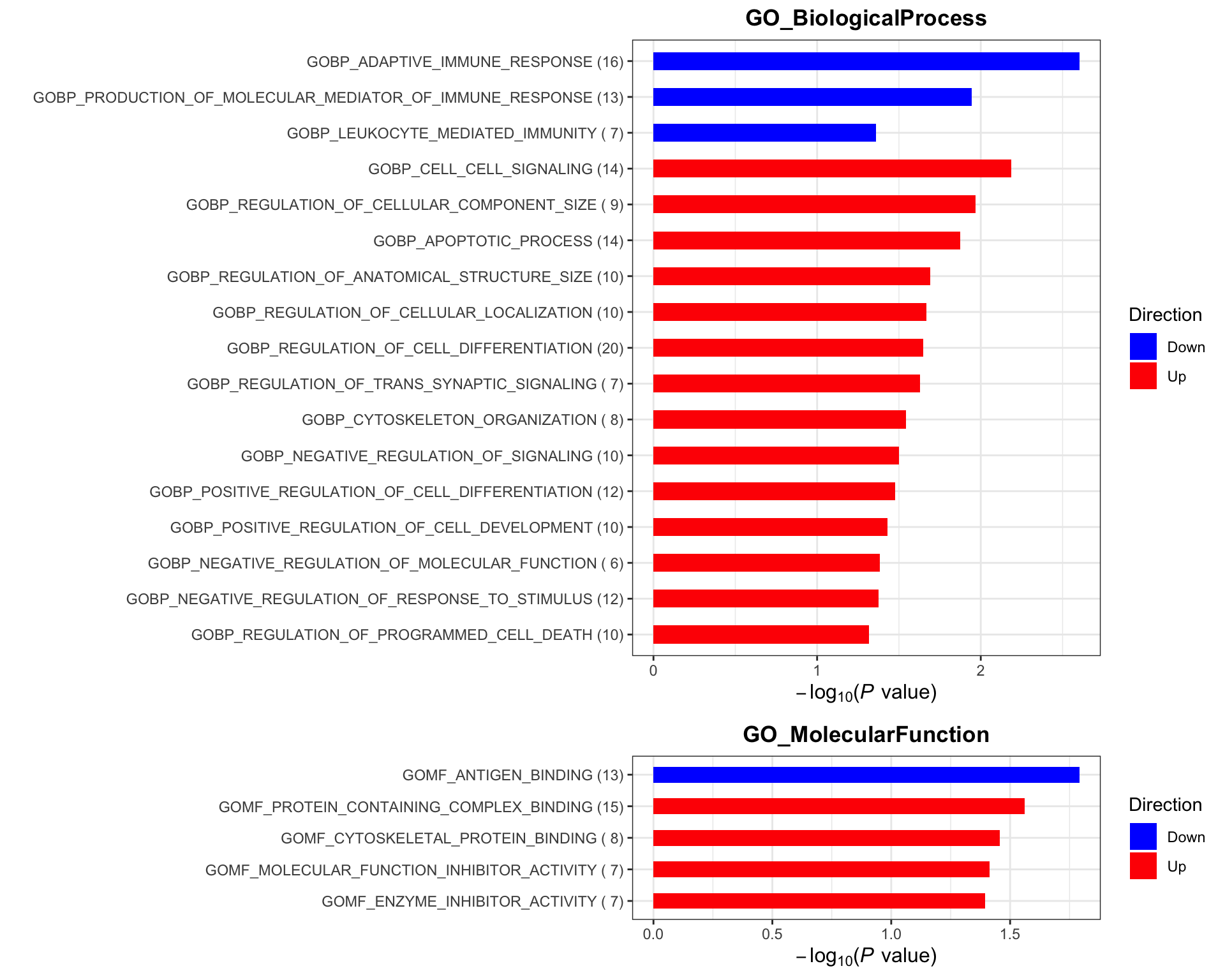
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.1
[3] dplyr_1.1.4.9000 purrr_0.3.4
[5] readr_2.1.2 tidyr_1.2.0
[7] tibble_3.2.1 ggplot2_3.4.1
[9] tidyverse_1.3.2 limma_3.52.2
[11] SummarizedExperiment_1.26.1 Biobase_2.56.0
[13] GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[15] IRanges_2.30.0 S4Vectors_0.34.0
[17] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[19] matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] utf8_1.2.4 shinydashboard_0.7.2 tidyselect_1.2.1
[4] RSQLite_2.2.15 AnnotationDbi_1.58.0 htmlwidgets_1.5.4
[7] grid_4.2.0 BiocParallel_1.30.3 maxstat_0.7-25
[10] munsell_0.5.0 codetools_0.2-18 DT_0.23
[13] withr_3.0.0 colorspace_2.0-3 highr_0.9
[16] knitr_1.39 rstudioapi_0.13 ggsignif_0.6.3
[19] labeling_0.4.2 git2r_0.30.1 slam_0.1-50
[22] GenomeInfoDbData_1.2.8 KMsurv_0.1-5 bit64_4.0.5
[25] farver_2.1.1 rprojroot_2.0.3 vctrs_0.6.5
[28] generics_0.1.3 TH.data_1.1-1 xfun_0.31
[31] sets_1.0-21 R6_2.5.1 ggbeeswarm_0.6.0
[34] locfit_1.5-9.6 bitops_1.0-7 cachem_1.0.6
[37] fgsea_1.22.0 DelayedArray_0.22.0 assertthat_0.2.1
[40] promises_1.2.0.1 scales_1.2.0 multcomp_1.4-19
[43] googlesheets4_1.0.0 beeswarm_0.4.0 gtable_0.3.0
[46] sva_3.44.0 sandwich_3.0-2 workflowr_1.7.0
[49] rlang_1.1.3 genefilter_1.78.0 splines_4.2.0
[52] rstatix_0.7.0 gargle_1.2.0 broom_1.0.0
[55] BiocManager_1.30.18 yaml_2.3.5 abind_1.4-5
[58] modelr_0.1.8 crosstalk_1.2.0 backports_1.4.1
[61] httpuv_1.6.6 tools_4.2.0 relations_0.6-12
[64] ellipsis_0.3.2 gplots_3.1.3 jquerylib_0.1.4
[67] Rcpp_1.0.9 visNetwork_2.1.0 zlibbioc_1.42.0
[70] RCurl_1.98-1.7 ggpubr_0.4.0 cowplot_1.1.1
[73] zoo_1.8-10 haven_2.5.0 cluster_2.1.3
[76] exactRankTests_0.8-35 fs_1.5.2 magrittr_2.0.3
[79] data.table_1.14.8 reprex_2.0.1 survminer_0.4.9
[82] googledrive_2.0.0 mvtnorm_1.1-3 hms_1.1.1
[85] shinyjs_2.1.0 mime_0.12 evaluate_0.15
[88] xtable_1.8-4 XML_3.99-0.10 readxl_1.4.0
[91] gridExtra_2.3 compiler_4.2.0 KernSmooth_2.23-20
[94] crayon_1.5.2 htmltools_0.5.4 mgcv_1.8-40
[97] later_1.3.0 tzdb_0.3.0 lubridate_1.8.0
[100] DBI_1.1.3 dbplyr_2.2.1 MASS_7.3-58
[103] jyluMisc_0.1.5 BiocStyle_2.24.0 Matrix_1.5-4
[106] car_3.1-0 cli_3.6.2 marray_1.74.0
[109] parallel_4.2.0 igraph_1.3.4 pkgconfig_2.0.3
[112] km.ci_0.5-6 piano_2.12.0 xml2_1.3.3
[115] annotate_1.74.0 vipor_0.4.5 bslib_0.4.1
[118] XVector_0.36.0 drc_3.0-1 rvest_1.0.2
[121] digest_0.6.30 Biostrings_2.64.0 rmarkdown_2.14
[124] cellranger_1.1.0 fastmatch_1.1-3 survMisc_0.5.6
[127] edgeR_3.38.1 shiny_1.7.4 gtools_3.9.3
[130] lifecycle_1.0.4 nlme_3.1-158 jsonlite_1.8.3
[133] carData_3.0-5 fansi_1.0.6 pillar_1.9.0
[136] lattice_0.20-45 KEGGREST_1.36.3 fastmap_1.1.0
[139] httr_1.4.3 plotrix_3.8-2 survival_3.4-0
[142] glue_1.7.0 png_0.1-7 bit_4.0.4
[145] stringi_1.7.8 sass_0.4.2 blob_1.2.3
[148] caTools_1.18.2 memoise_2.0.1