Build machine learning model for predicting treatment outcome, delta_UEMS
Junyan Lu
17 May 2024
Last updated: 2024-05-17
Checks: 5 1
Knit directory:
SpinalCord_proteomics/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20221110) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Preparing dataset
Subset and removed unwanted variations
Subset
seSub <- prepareProt(seProt, filterCondi = list(Treatment = "1"), perNA = 1)[1] "Number of proteins: 473, number of samples: 184"sva
mod <- model.matrix(~ Visit + AIS + UEMS + delta_UEMS + nodeGroup, colData(seSub))
exprMat <- assays(seSub)[[2]]
svaObj <- sva::sva(exprMat, mod)Number of significant surrogate variables is: 10
Iteration (out of 5 ):1 2 3 4 5 assays(seSub)[[1]] <- limma::removeBatchEffect(assay(seSub), covariates = svaObj$sv)
assays(seSub)[[2]] <- limma::removeBatchEffect(assays(seSub)[[2]], covariates = svaObj$sv)Prepare each proteomic dataset for comparing
Data set for individual time point
protV3 <- prepareProt(seProt, filterCondi = list(Treatment = "1", Visit = 3), perNA = 0.5)[1] "Number of proteins: 378, number of samples: 66"colnames(protV3) <- protV3$PSN
protV8 <- prepareProt(seProt, filterCondi = list(Treatment = "1", Visit = 8), perNA = 0.5)[1] "Number of proteins: 374, number of samples: 61"colnames(protV8) <- protV8$PSN
protV10 <- prepareProt(seProt, filterCondi = list(Treatment = "1", Visit = 10), perNA = 0.5)[1] "Number of proteins: 374, number of samples: 57"colnames(protV10) <- protV10$PSNCalculate the protein expression difference before and after treatment (V8-V3)
overPat <- intersect(protV8$PSN, protV3$PSN)
overProt <- intersect(rownames(protV3), rownames(protV8))
protSub.before <- protV3[overProt,match(overPat, protV3$PSN)]
protSub.after <- protV8[overProt,match(overPat, protV8$PSN)]
colnames(protSub.after) <- overPat
colnames(protSub.before) <- overPat
protDiff38 <- protSub.before
assays(protDiff38)[[1]] <- assays(protSub.after)[[1]] - assays(protSub.before)[[1]]
assays(protDiff38)[[2]] <- assays(protSub.after)[[2]] - assays(protSub.before)[[2]]
#remove feature with too many missings
#protSub <- protSub[rowSums(is.na(assay(protSub)))/ncol(protSub) < 0.5,]
print("How many proteins and samples")[1] "How many proteins and samples"dim(protDiff38)[1] 372 61Generate training dataset
For individual data, pre-filtering by association test, raw P < 0.1, to reduce the dimentions
pCut = 0.05
allTrainData <- list(
V3 = list(y = protV3$delta_UEMS,
X = assays(protV3)[[2]][rownames(protV3) %in% filter(allResList$corrOutcome_baseline_treated, pval <= pCut)$symbol,]),
V8 = list(y = protV8$delta_UEMS,
X = assays(protV8)[[2]][rownames(protV8) %in% filter(allResList$corrOutcome_visit8_treated, pval <= pCut)$symbol,]),
#V10 = list(y = protV10$delta_UEMS,
# X = assays(protV10)[[2]][rownames(protV10) %in% filter(allResList$corrOutcome_visit10_treated, pval <= pCut)$symbol,]),
diff38 = list(y = protDiff38$delta_UEMS,
X = assays(protDiff38)[[2]][rownames(protDiff38) %in% filter(allResList$corrOutcome_diff83_treated, pval <= pCut)$symbol,])
)Combined data
overPat <- jyluMisc::overlap(colnames(protV3), colnames(protV8), colnames(protV10), colnames(protDiff38))
comMat <- lapply(names(allTrainData), function(n) {
exprMat <- allTrainData[[n]]$X[,overPat]
rownames(exprMat) <- paste0(n,"_",rownames(exprMat))
exprMat
}) %>% do.call(rbind,.)
allTrainData[["combine"]] <- list(y = protDiff38[,overPat]$delta_UEMS, X = comMat[,overPat])Use random lasso to rank features by their importance
source("../code/Random_lasso.R")Calculate feature importance patth by random lasso
set.seed(2024)
rndLassoRes <- list()
rndRes <- lapply(names(allTrainData), function(n) {
X <- scale(t(allTrainData[[n]]$X))
y <- allTrainData[[n]]$y
res <- featurePath(X, y, sampleFraction = 0.5, weakness =0.2, nPerm = 1000,
typePerm = "standard", lambda = seq(1,0.01,length.out = 100))
tibble(feature = rownames(res$freqMat),
importance = rowMeans(res$freqMat),
set = n)
}) %>% bind_rows()
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
List of the top 30 most important features
pList <- lapply(unique(rndRes$set), function(s) {
eachTab <- filter(rndRes, set == s) %>%
arrange(desc(importance)) %>%
slice_head(n=30) %>%
mutate(feature = factor(feature, levels = feature))
ggplot(eachTab, aes(x=feature, y=importance)) +
geom_bar(stat ="identity") +
ggtitle(s) +
theme_bw() +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0),
plot.title = element_text(face ="bold", size=10, hjust = 0.5))
})
cowplot::plot_grid(plotlist= pList, ncol=1)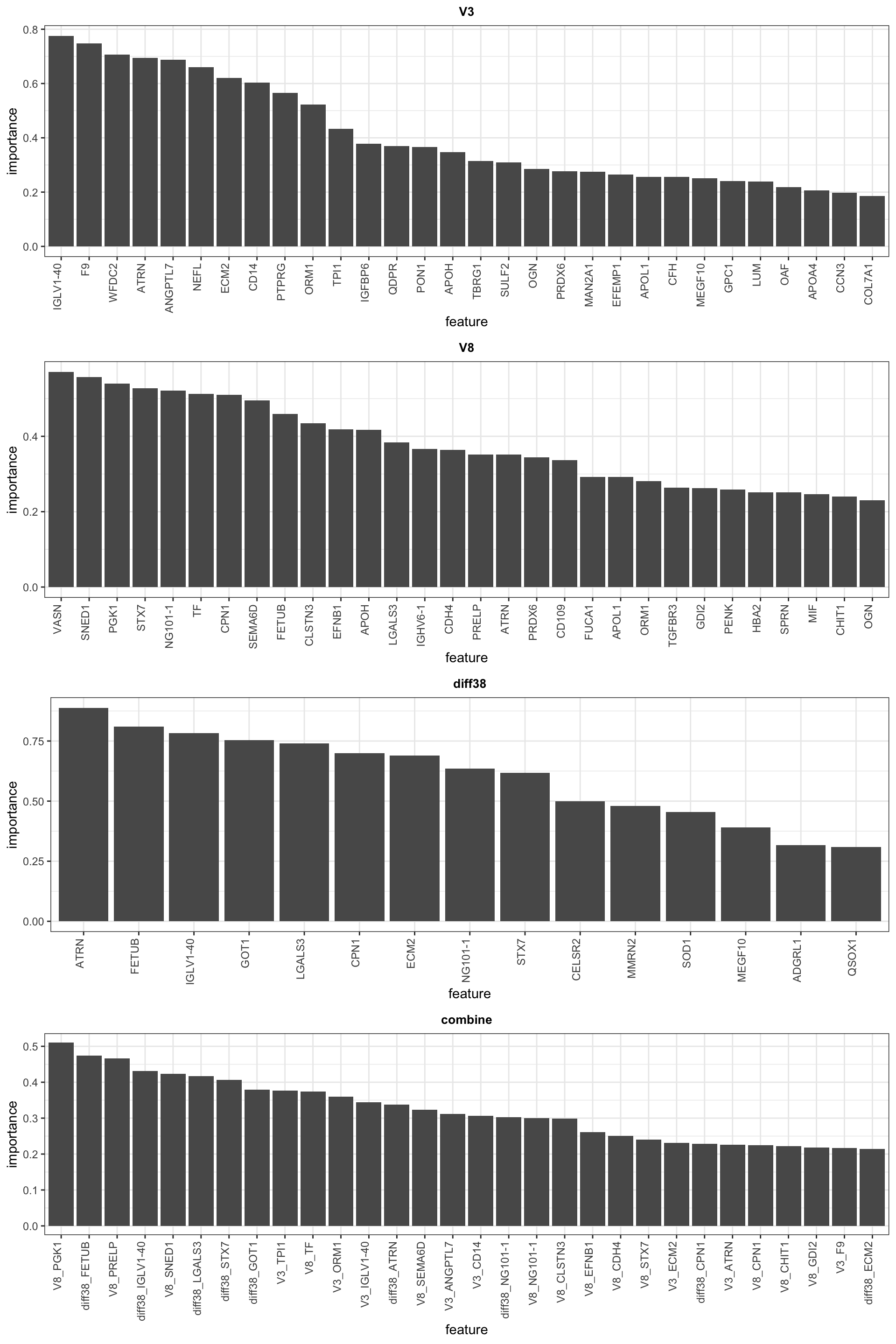
Test the performance of using different number of features
set.seed(2024)
featureNum <- c(3,5, seq(10,20),30)
allCompareRes <- lapply(unique(names(allTrainData)), function(n) {
eachCompareRes <- lapply(featureNum, function(m) {
seleFeature <- filter(rndRes, set == n) %>%
slice_max(importance, n=m) %>% pull(feature)
X <- scale(t(allTrainData[[n]]$X))[,seleFeature]
y <- allTrainData[[n]]$y
perf <- testModel(X, y, repeats = 100, testRatio = 0.3)
tibble(set = n, num = m,
meanR2 = mean(perf, na.rm=TRUE),
sdR2 = sd(perf, na.rm=TRUE),
CI = 1.96*sdR2/sqrt(length(perf)))
}) %>% bind_rows()
}) %>% bind_rows()
Warning: The above code chunk cached its results, but
it won’t be re-run if previous chunks it depends on are updated. If you
need to use caching, it is highly recommended to also set
knitr::opts_chunk$set(autodep = TRUE) at the top of the
file (in a chunk that is not cached). Alternatively, you can customize
the option dependson for each individual chunk that is
cached. Using either autodep or dependson will
remove this warning. See the
knitr cache options for more details.
ggplot(allCompareRes, aes(x=factor(num), y=meanR2, fill = set)) +
geom_bar(stat="identity") +
geom_errorbar(aes(ymax = meanR2 + CI, ymin = meanR2-CI), width=0.5) +
facet_wrap(~set, scale = "free_x") +
theme(legend.position = "none") +
xlab("Number of protein markers") + ylab("Variance explained") +
theme_bw() + theme(legend.position = "none")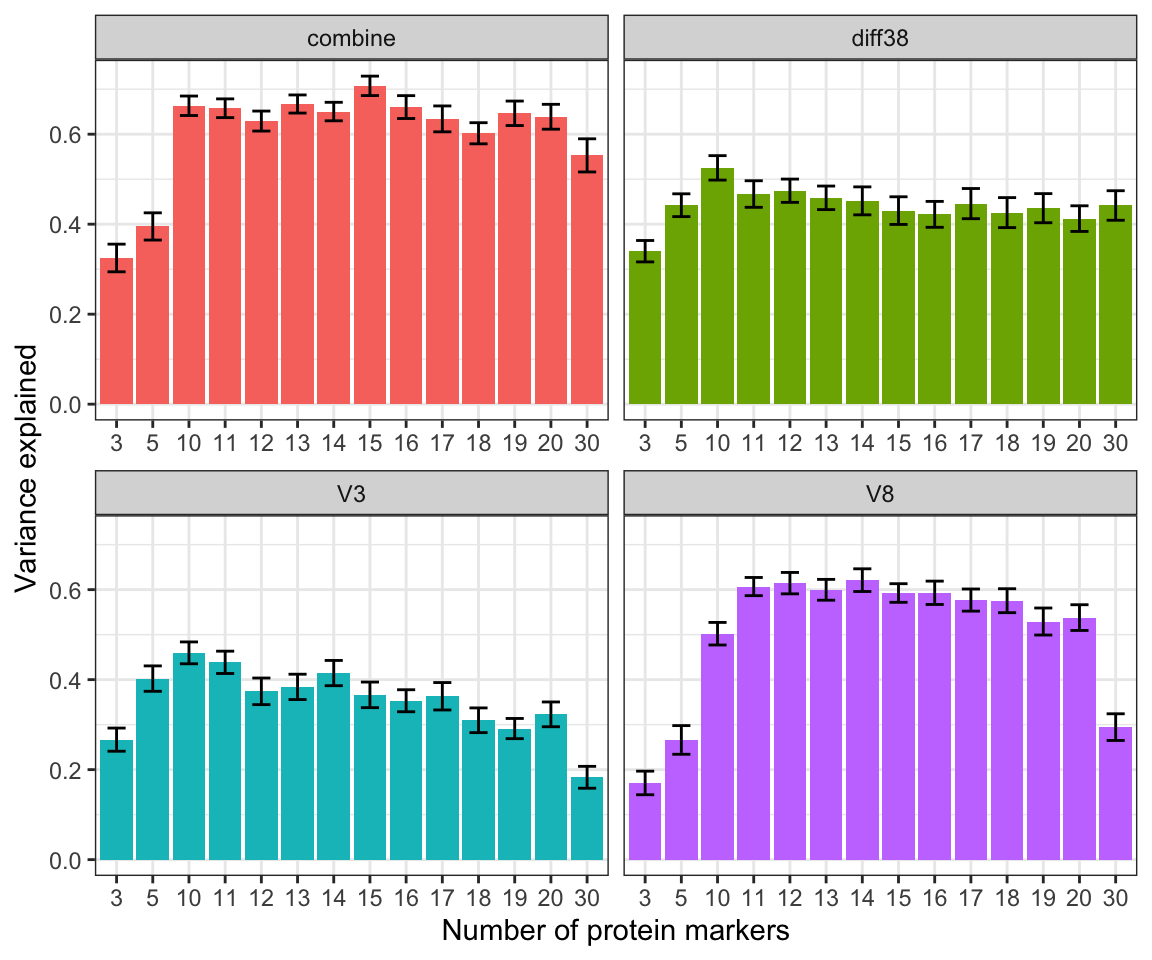 The higher variance explained, the better the models
are This graph shows that the best performing model are
1) Using 15 proteins from the combined V3 and V8 proteomics. 2) Using
proteins from the V8 proteomic dataset (slightly worse than the first
mdoel) If we only want to use the baseline for
prediction, 10 features from the V3 dataset performs the
best
The higher variance explained, the better the models
are This graph shows that the best performing model are
1) Using 15 proteins from the combined V3 and V8 proteomics. 2) Using
proteins from the V8 proteomic dataset (slightly worse than the first
mdoel) If we only want to use the baseline for
prediction, 10 features from the V3 dataset performs the
best
Visualize the 15 features from the combined dataset
seleFeature <- filter(rndRes, set == "combine") %>%
slice_max(importance, n=15) %>% pull(feature)
#predict the score
scoreList <- list()
X <- t(allTrainData[["combine"]]$X)[,seleFeature]
y <- allTrainData[["combine"]]$y
seleModel <- glmnet(X, y, lambda =0)
y.pred <- predict(seleModel, newx = X)[,1]
scoreList[["combine_15"]] <- y.pred
plotMat <- allTrainData$combine$X[seleFeature,]
annoCol <- data.frame(row.names = colnames(plotMat), delta_UEMS = allTrainData$combine$y,
proteinScore = y.pred,
nodeGroup = protDiff38[,colnames(plotMat)]$nodeGroup)
annoCol <- annoCol[order(annoCol$delta_UEMS),,drop=FALSE]
plotMat <- plotMat[,rownames(annoCol)]
pheatmap::pheatmap(plotMat, annotation_col = annoCol, cluster_rows = TRUE, cluster_cols = FALSE, scale = "row",
clustering_method = "ward.D2",
color = colorRampPalette(c("blue", "white", "red"))(100))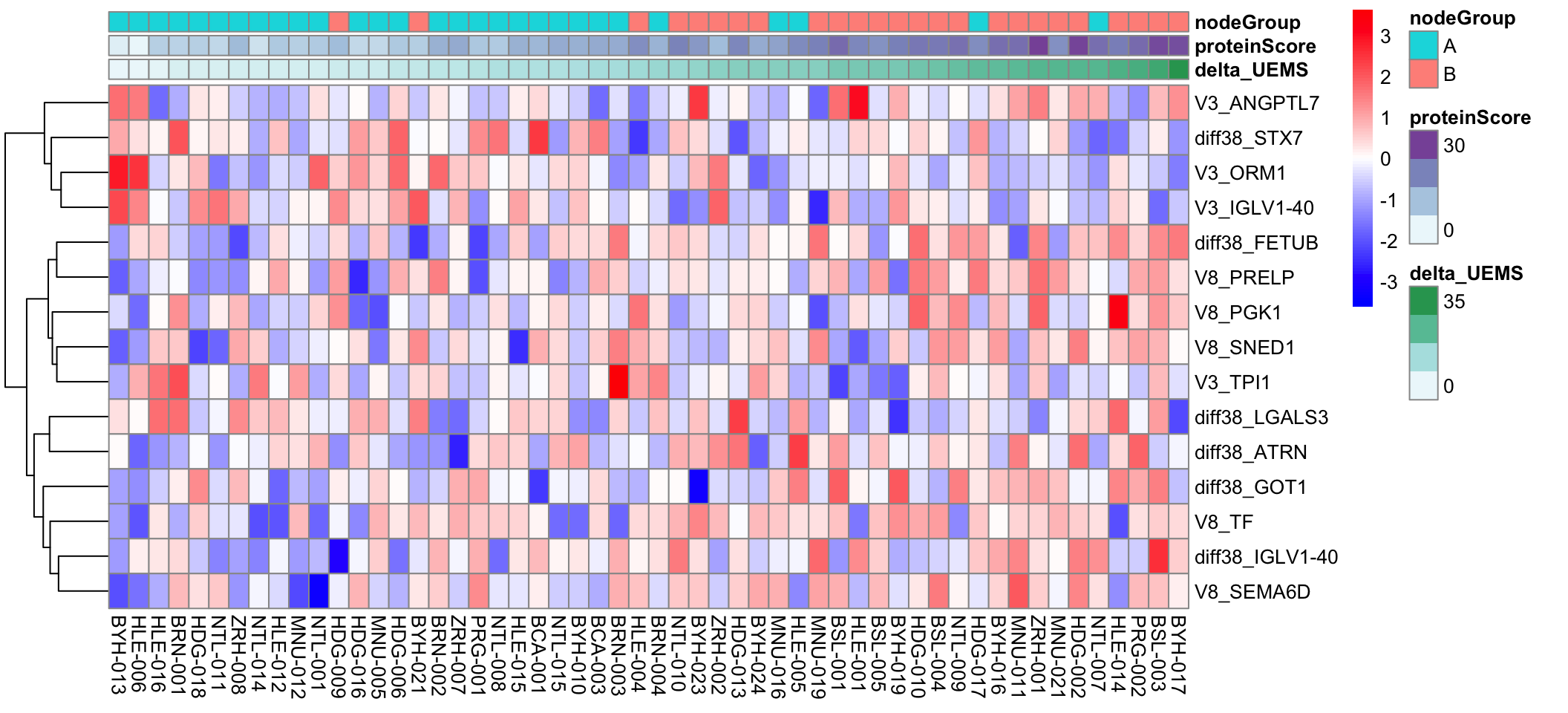
Visualize the 14 features from the V8 dataset
seleFeature <- filter(rndRes, set == "V8") %>%
slice_max(importance, n=14) %>% pull(feature)
#predict the score
X <- t(allTrainData[["V8"]]$X)[,seleFeature]
y <- allTrainData[["V8"]]$y
seleModel <- glmnet(X, y, lambda =0)
y.pred <- predict(seleModel, newx = X)[,1]
scoreList[["V8_14"]] <- y.pred
plotMat <- allTrainData$V8$X[seleFeature,]
annoCol <- data.frame(row.names = colnames(plotMat), delta_UEMS = allTrainData$V8$y,
proteinScore = y.pred,
nodeGroup = protV8[,colnames(plotMat)]$nodeGroup)
annoCol <- annoCol[order(annoCol$delta_UEMS),,drop=FALSE]
plotMat <- plotMat[,rownames(annoCol)]
pheatmap::pheatmap(plotMat, annotation_col = annoCol, cluster_rows = TRUE, cluster_cols = FALSE, scale = "row", clustering_method = "ward.D2",
color = colorRampPalette(c("blue", "white", "red"))(100))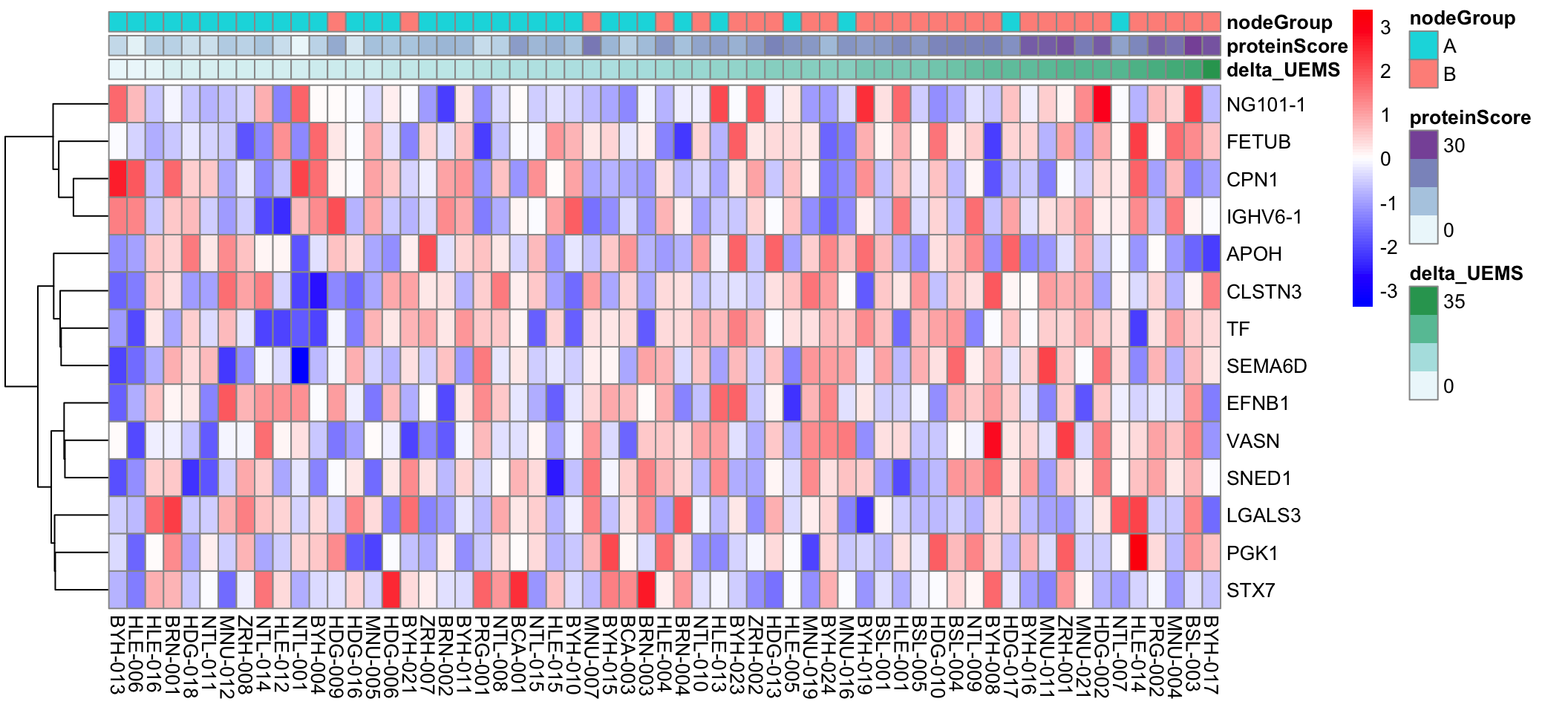
Visualize the 10 features from the V3 dataset
seleFeature <- filter(rndRes, set == "V3") %>%
slice_max(importance, n=10) %>% pull(feature)
#predict the score
X <- t(allTrainData[["V3"]]$X)[,seleFeature]
y <- allTrainData[["V3"]]$y
seleModel <- glmnet(X, y, lambda =0)
y.pred <- predict(seleModel, newx = X)[,1]
scoreList[["V3_10"]] <- y.pred
plotMat <- allTrainData$V3$X[seleFeature,]
annoCol <- data.frame(row.names = colnames(plotMat), delta_UEMS = allTrainData$V3$y,
proteinScore = y.pred,
nodeGroup = protV3[,colnames(plotMat)]$nodeGroup)
annoCol <- annoCol[order(annoCol$delta_UEMS),,drop=FALSE]
plotMat <- plotMat[,rownames(annoCol)]
pheatmap::pheatmap(plotMat, annotation_col = annoCol, cluster_rows = TRUE, cluster_cols = FALSE, scale = "row",
clustering_method = "ward.D2",
color = colorRampPalette(c("blue", "white", "red"))(100))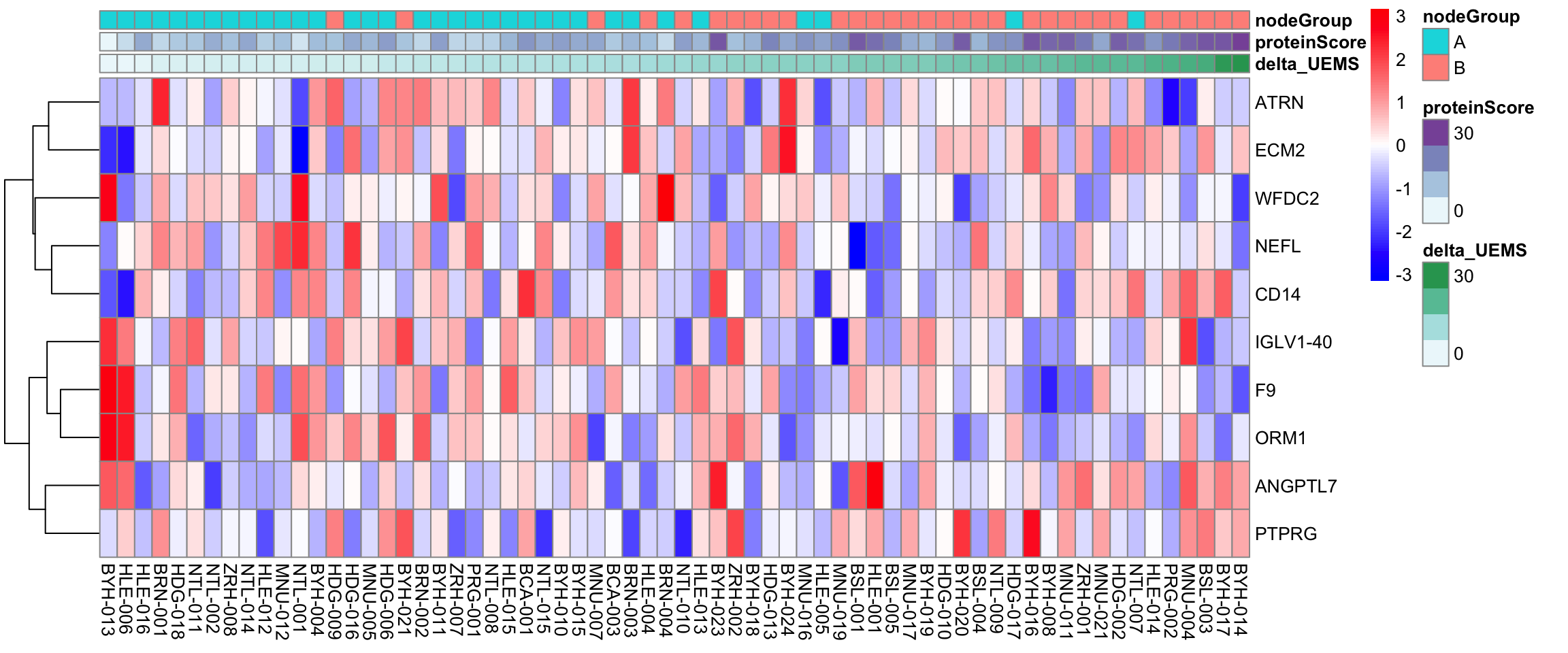
Test the score performance in multi-variate models including other clinical paramters
Prepare a table for other clinical parameters
Those parameter will be included: Age, Sex, UEMS at Visit 3, AIS, random node group (A or B)
patAnno <- colData(seProt) %>% as_tibble() %>% filter(Treatment =="1")
scaleCol <- function(x){
return((x-mean(x,na.rm=TRUE))/sd(x, na.rm=TRUE))
}
clinicTab <- arrange(patAnno, Visit) %>% distinct(PSN,.keep_all = TRUE) %>%
select(PSN, SEX, AGE, AIS, nodeGroup, delta_UEMS) %>%
mutate_if(is.numeric, scaleCol)
uemsTab <- distinct(patAnno, PSN, Visit, UEMS) %>%
mutate(Visit = paste0("UEMS_V",Visit)) %>%
pivot_wider(names_from = Visit, values_from = UEMS) %>%
#mutate(UEMS_diff38 = UEMS_V8/UEMS_V3) %>%
select(PSN, UEMS_V3)
clinicTab <- left_join(clinicTab, uemsTab, by = "PSN") %>%
mutate_if(is.character, as.factor) %>%
mutate(PSN = as.character(PSN))Test performance of model with only clinical parameter, protein score and clinical paremeter + protein score
With the combine_15 protein score
Prediction accuracy
allModels <- list()
allModels[["combine"]] <- clinicTab %>% mutate(proteinScore = scoreList$combine_15[PSN]) %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyClinic"]] <- clinicTab %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyProtein"]] <- allModels$combine[,c("delta_UEMS","proteinScore")]
plotResTab <- lapply(names(allModels), function(nn) {
eachModel <- allModels[[nn]]
res <- summary(lm(delta_UEMS~. ,eachModel))
tibble(model = nn, R2 = res$adj.r.squared)
}) %>% bind_rows()
ggplot(plotResTab, aes(x=model, y=R2, fill = model)) +
geom_bar(stat = "identity") +
coord_flip() +
ylim(0,1) +
theme_bw() + theme(legend.position = "none") +
ylab("Variance explained")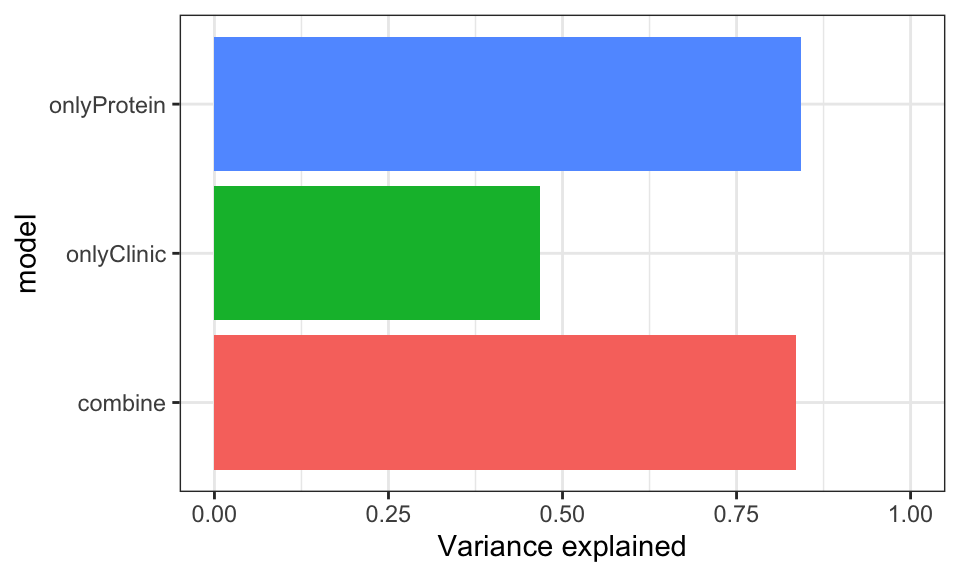 The higher the variance explained, the better the models
are
The higher the variance explained, the better the models
are
Forest plot for feature importance and significance
res <- summary(lm(delta_UEMS~. ,allModels$combine))
plotForest(res)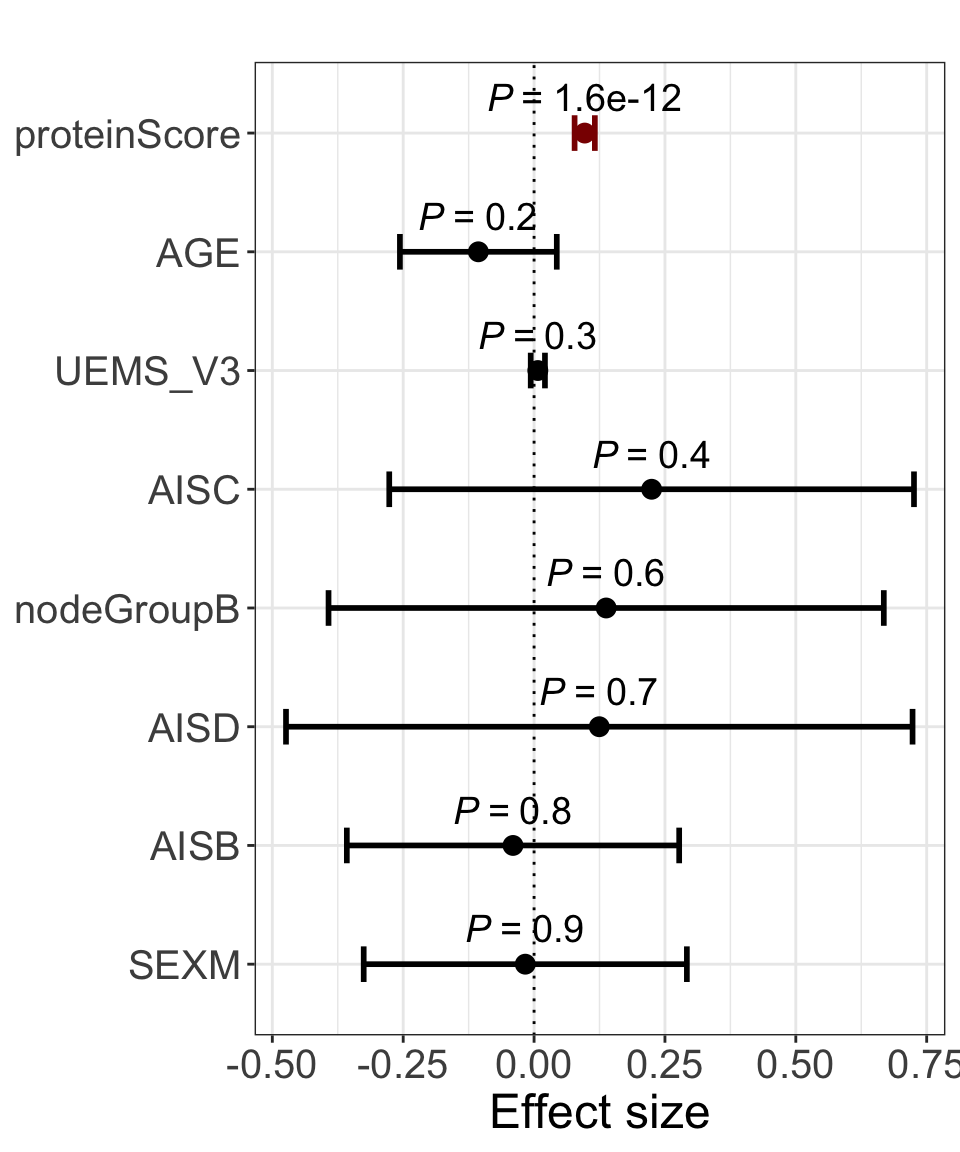
With the V8_14 score
Prediction accuracy
allModels <- list()
allModels[["combine"]] <- clinicTab %>% mutate(proteinScore = scoreList$V8_14[PSN]) %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyClinic"]] <- clinicTab %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyProtein"]] <- allModels$combine[,c("delta_UEMS","proteinScore")]
plotResTab <- lapply(names(allModels), function(nn) {
eachModel <- allModels[[nn]]
res <- summary(lm(delta_UEMS~. ,eachModel))
tibble(model = nn, R2 = res$adj.r.squared)
}) %>% bind_rows()
ggplot(plotResTab, aes(x=model, y=R2, fill = model)) +
geom_bar(stat = "identity") +
coord_flip() +
theme_bw() + theme(legend.position = "none") +
ylim(0,1)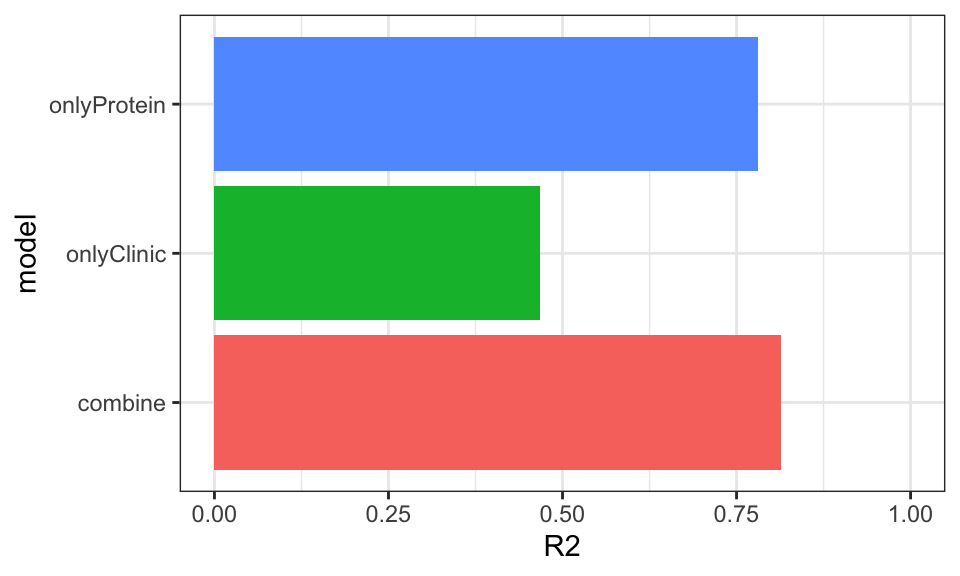
Forest plot for feature importance and significance
res <- summary(lm(delta_UEMS~. ,allModels$combine))
plotForest(res)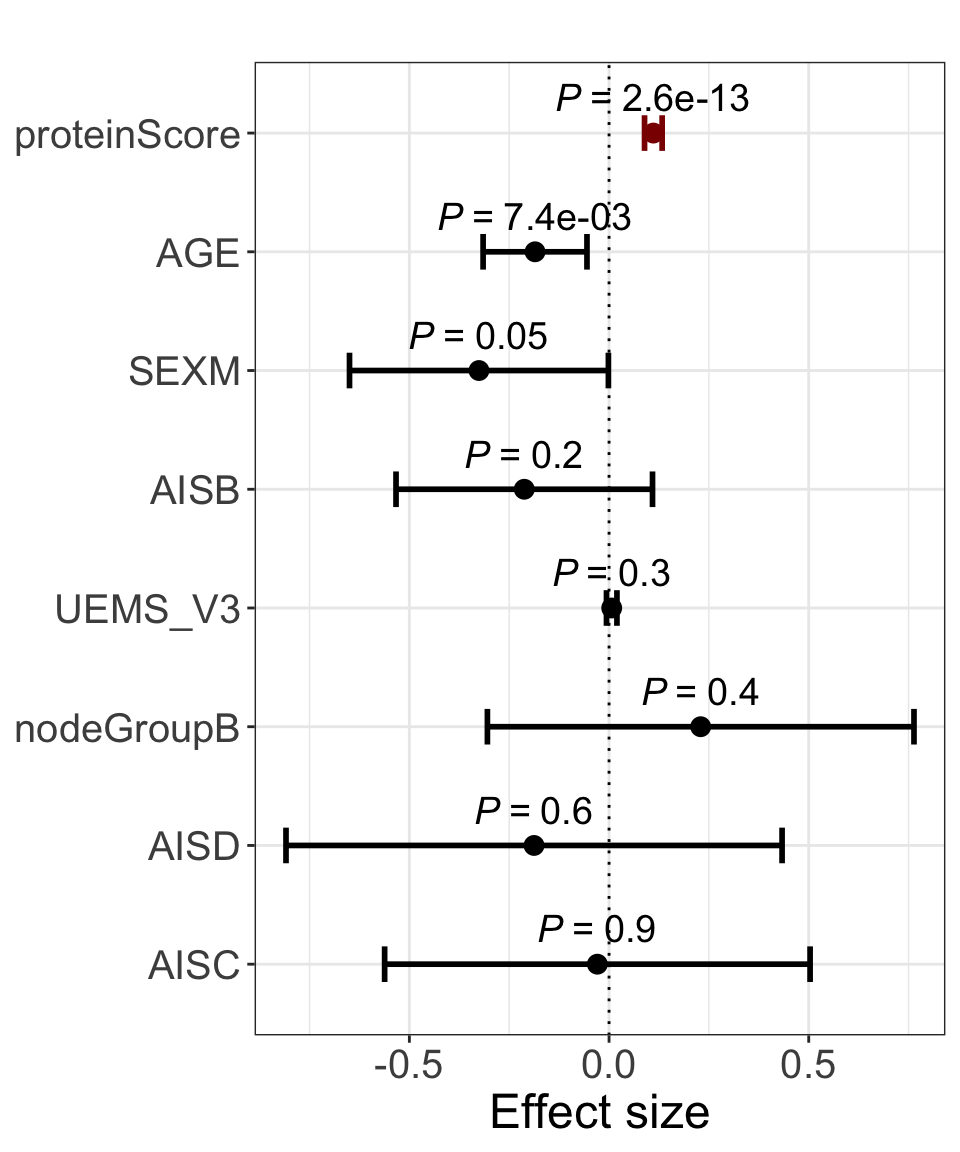
Forest plot for only clinical variables
res <- summary(lm(delta_UEMS~. ,allModels$onlyClinic))
plotForest(res)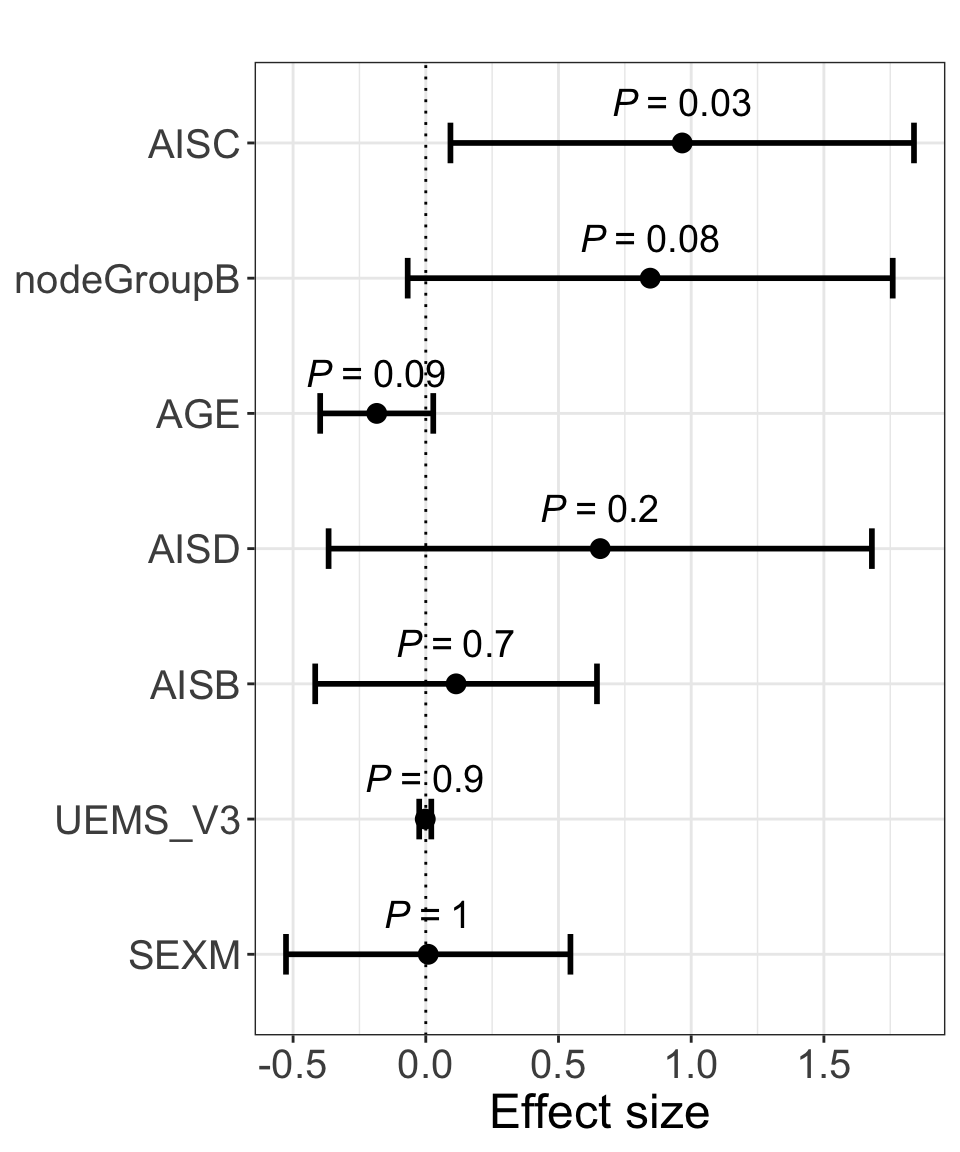
With the V3_10 score
Prediction accuracy
allModels <- list()
allModels[["combine"]] <- clinicTab %>% mutate(proteinScore = scoreList$V3_10[PSN]) %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyClinic"]] <- clinicTab %>%
column_to_rownames("PSN") %>% data.frame()
allModels[["onlyProtein"]] <- allModels$combine[,c("delta_UEMS","proteinScore")]
plotResTab <- lapply(names(allModels), function(nn) {
eachModel <- allModels[[nn]]
res <- summary(lm(delta_UEMS~. ,eachModel))
tibble(model = nn, R2 = res$adj.r.squared)
}) %>% bind_rows()
ggplot(plotResTab, aes(x=model, y=R2, fill = model)) +
geom_bar(stat = "identity") +
coord_flip() +
theme_bw() + theme(legend.position = "none") +
ylim(0,1)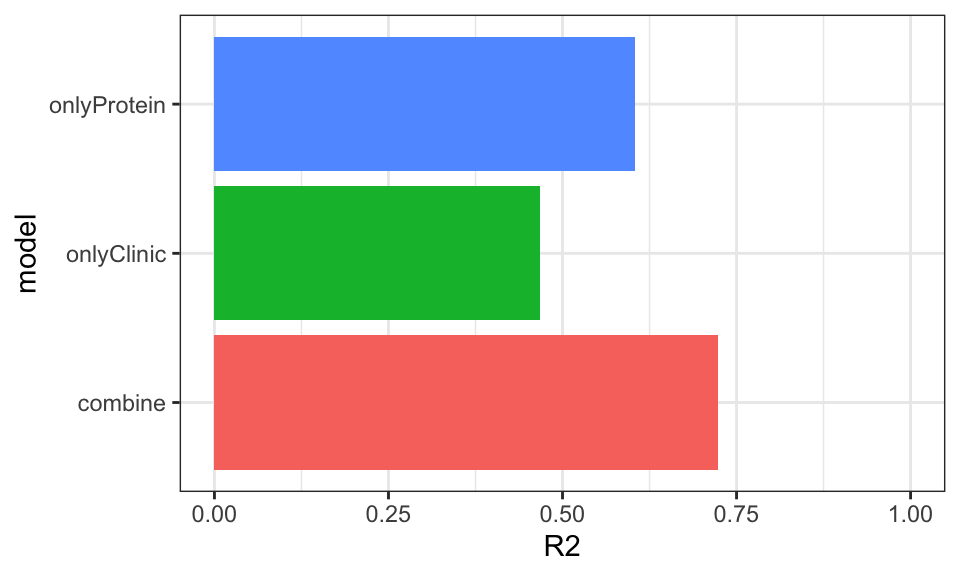
Forest plot for feature importance and significance
res <- summary(lm(delta_UEMS~. ,allModels$combine))
plotForest(res)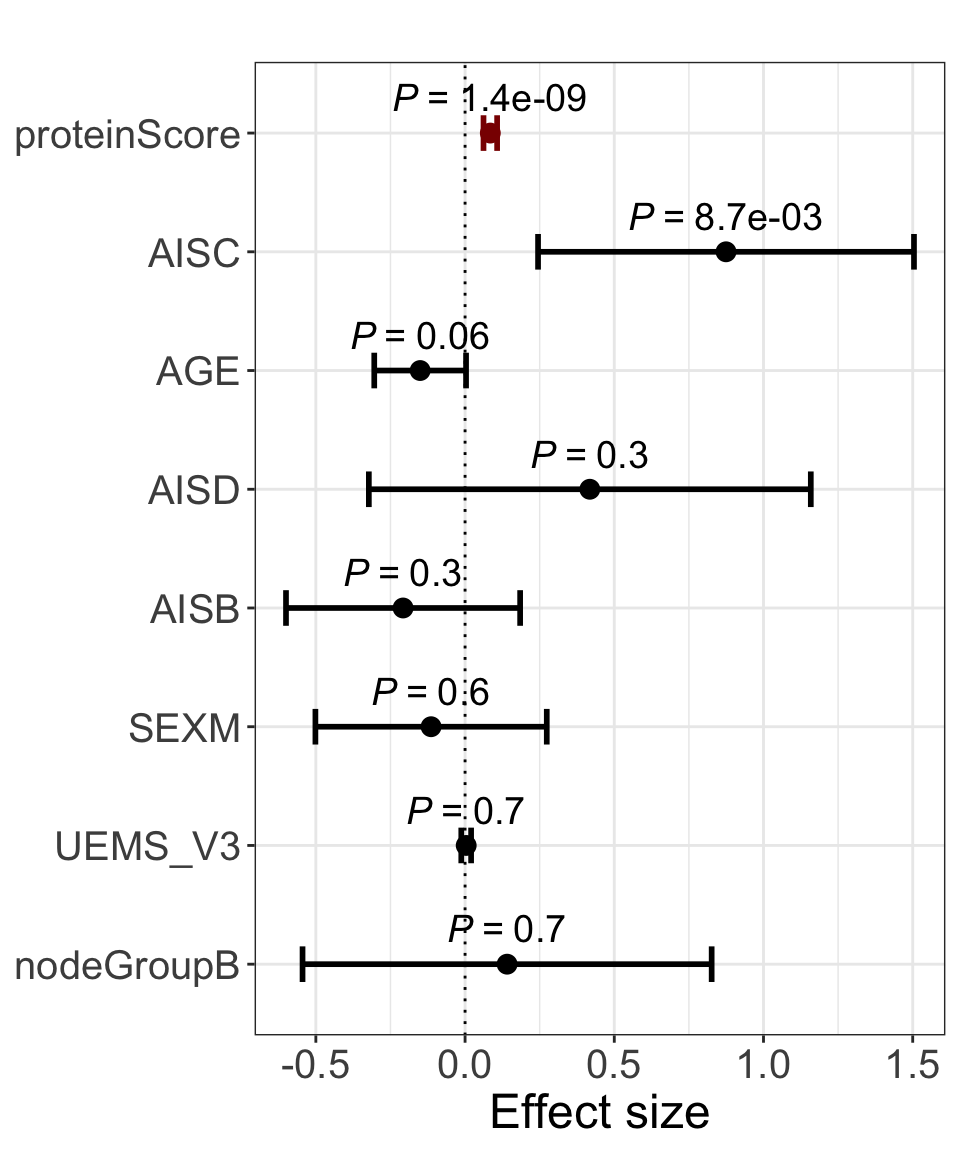
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] forcats_0.5.1 stringr_1.4.1
[3] dplyr_1.1.4.9000 purrr_0.3.4
[5] readr_2.1.2 tidyr_1.2.0
[7] tibble_3.2.1 ggplot2_3.4.1
[9] tidyverse_1.3.2 glmnet_4.1-4
[11] Matrix_1.5-4 sva_3.44.0
[13] BiocParallel_1.30.3 genefilter_1.78.0
[15] mgcv_1.8-40 nlme_3.1-158
[17] limma_3.52.2 SummarizedExperiment_1.26.1
[19] Biobase_2.56.0 GenomicRanges_1.48.0
[21] GenomeInfoDb_1.32.2 IRanges_2.30.0
[23] S4Vectors_0.34.0 BiocGenerics_0.42.0
[25] MatrixGenerics_1.8.1 matrixStats_0.62.0
loaded via a namespace (and not attached):
[1] utf8_1.2.4 shinydashboard_0.7.2 tidyselect_1.2.1
[4] RSQLite_2.2.15 AnnotationDbi_1.58.0 htmlwidgets_1.5.4
[7] grid_4.2.0 maxstat_0.7-25 munsell_0.5.0
[10] codetools_0.2-18 DT_0.23 withr_3.0.0
[13] colorspace_2.0-3 highr_0.9 knitr_1.39
[16] rstudioapi_0.13 ggsignif_0.6.3 labeling_0.4.2
[19] git2r_0.30.1 slam_0.1-50 GenomeInfoDbData_1.2.8
[22] KMsurv_0.1-5 bit64_4.0.5 farver_2.1.1
[25] pheatmap_1.0.12 rprojroot_2.0.3 vctrs_0.6.5
[28] generics_0.1.3 TH.data_1.1-1 xfun_0.31
[31] sets_1.0-21 R6_2.5.1 locfit_1.5-9.6
[34] bitops_1.0-7 cachem_1.0.6 fgsea_1.22.0
[37] DelayedArray_0.22.0 assertthat_0.2.1 promises_1.2.0.1
[40] scales_1.2.0 multcomp_1.4-19 googlesheets4_1.0.0
[43] gtable_0.3.0 sandwich_3.0-2 workflowr_1.7.0
[46] rlang_1.1.3 splines_4.2.0 rstatix_0.7.0
[49] gargle_1.2.0 broom_1.0.0 BiocManager_1.30.18
[52] yaml_2.3.5 abind_1.4-5 modelr_0.1.8
[55] backports_1.4.1 httpuv_1.6.6 tools_4.2.0
[58] relations_0.6-12 ellipsis_0.3.2 gplots_3.1.3
[61] RColorBrewer_1.1-3 jquerylib_0.1.4 Rcpp_1.0.9
[64] visNetwork_2.1.0 zlibbioc_1.42.0 RCurl_1.98-1.7
[67] ggpubr_0.4.0 cowplot_1.1.1 zoo_1.8-10
[70] haven_2.5.0 cluster_2.1.3 exactRankTests_0.8-35
[73] fs_1.5.2 magrittr_2.0.3 data.table_1.14.8
[76] reprex_2.0.1 survminer_0.4.9 googledrive_2.0.0
[79] mvtnorm_1.1-3 hms_1.1.1 shinyjs_2.1.0
[82] mime_0.12 evaluate_0.15 xtable_1.8-4
[85] XML_3.99-0.10 readxl_1.4.0 gridExtra_2.3
[88] shape_1.4.6 compiler_4.2.0 KernSmooth_2.23-20
[91] crayon_1.5.2 htmltools_0.5.4 later_1.3.0
[94] tzdb_0.3.0 lubridate_1.8.0 DBI_1.1.3
[97] dbplyr_2.2.1 MASS_7.3-58 jyluMisc_0.1.5
[100] BiocStyle_2.24.0 car_3.1-0 cli_3.6.2
[103] marray_1.74.0 parallel_4.2.0 igraph_1.3.4
[106] pkgconfig_2.0.3 km.ci_0.5-6 piano_2.12.0
[109] xml2_1.3.3 foreach_1.5.2 annotate_1.74.0
[112] bslib_0.4.1 XVector_0.36.0 drc_3.0-1
[115] rvest_1.0.2 digest_0.6.30 Biostrings_2.64.0
[118] rmarkdown_2.14 cellranger_1.1.0 fastmatch_1.1-3
[121] survMisc_0.5.6 edgeR_3.38.1 shiny_1.7.4
[124] gtools_3.9.3 lifecycle_1.0.4 jsonlite_1.8.3
[127] carData_3.0-5 fansi_1.0.6 pillar_1.9.0
[130] lattice_0.20-45 KEGGREST_1.36.3 fastmap_1.1.0
[133] httr_1.4.3 plotrix_3.8-2 survival_3.4-0
[136] glue_1.7.0 png_0.1-7 iterators_1.0.14
[139] bit_4.0.4 stringi_1.7.8 sass_0.4.2
[142] blob_1.2.3 caTools_1.18.2 memoise_2.0.1