Analysis of combinatorial effect using simple bliss model
Junyan Lu
2022-04-25
Last updated: 2022-09-02
Checks: 5 1
Knit directory: combiDLBCL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220425) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load libraries and datasets
Calculate CI for the combination of bases and combi drugs
Only CHP_Pola plates will be used
screenSub <- filter(screenData, Drug_B == "CHP_Pola")Get combination effect
comTab <- filter(screenSub, Drug_A.Conc >0, Drug_B.Conc >0) %>%
select(Name, Drug_A, Drug_A.Conc, Drug_B, Drug_B.Conc, normVal) %>%
dplyr::rename(viabObs = normVal)
drugATab <- filter(screenSub, Drug_A != "DMSO", Drug_B.Conc ==0) %>%
select(Name, Drug_A, Drug_A.Conc, normVal, Drug_A.ConcStep) %>%
dplyr::rename(viabA = normVal)
drugBTab <- filter(screenSub, Drug_A == "DMSO", Drug_B.Conc !=0) %>%
select(Name, Drug_B, Drug_B.Conc, normVal) %>%
dplyr::rename(viabB = normVal)synTab <- comTab %>% left_join(drugATab, by =c("Name","Drug_A","Drug_A.Conc")) %>%
left_join(drugBTab, by = c("Name","Drug_B","Drug_B.Conc")) %>%
mutate(viabExp = viabA*viabB) %>%
mutate(CI = viabObs-viabExp,
logCI = log10(viabObs/viabExp))Visualize CI for each drug and concentration
ggplot(synTab, aes(x=Drug_A.ConcStep, y=Drug_A, fill = CI)) +
geom_tile() +
facet_wrap(~Name) +
scale_fill_gradient2(low ="red",high="blue",mid="white")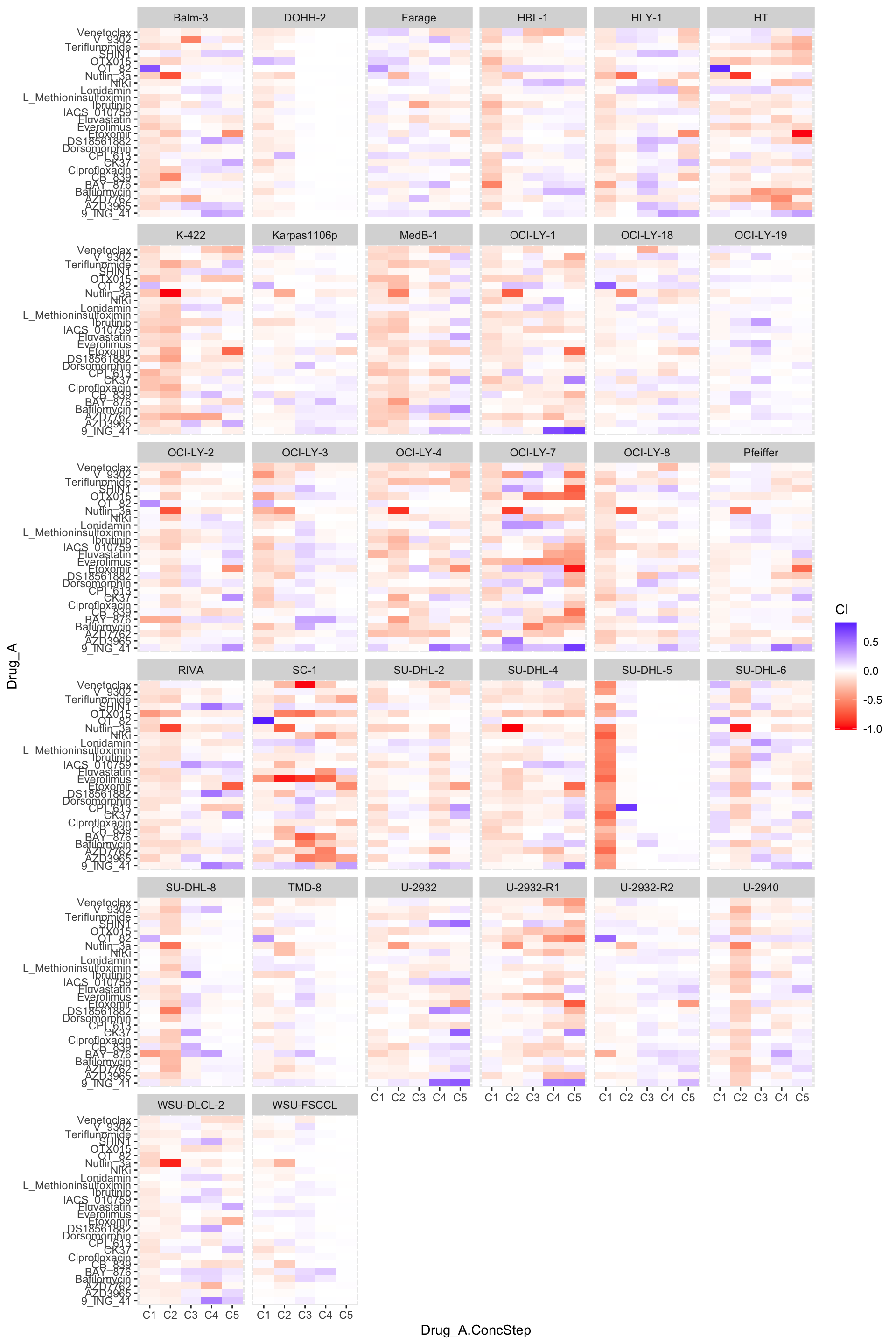
Summarise CI
Calculate synergistic and antagonistic effect separately, using a similar way as bayesyngergy package
sumSyn <- function(viabExp, viabObs) {
tab <- tibble(viabExp=viabExp, viabObs = viabObs) %>%
mutate(syn = min(0, viabObs - viabExp),
anta = max(0, viabObs - viabExp))
return(tibble(syn = sum(tab$syn), anta = sum(tab$anta)))
}
ciTabSum <- group_by(synTab, Name, Drug_A) %>% nest() %>%
mutate(res = map(data, ~sumSyn(.$viabExp, .$viabObs))) %>%
unnest(res) %>% select(-data)Visualization of synergistic and antagoistic effect
Scatter plot
Top 5% synergistics or antagonistic effect are labelled.
plotSynScatter(ciTabSum, 0.05)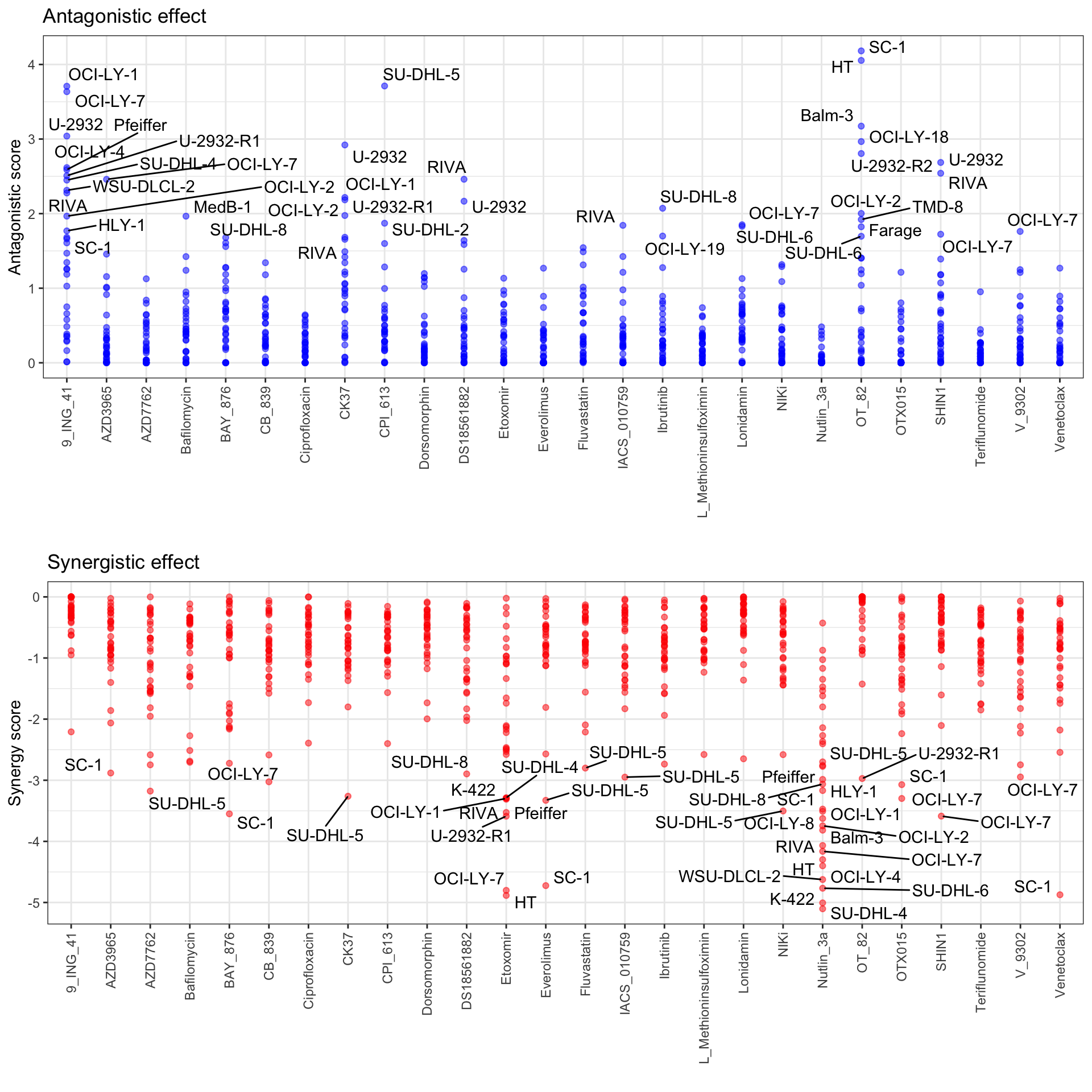
Matrix visualization
plotSynMatrix(ciTabSum, 0.1, 0.2)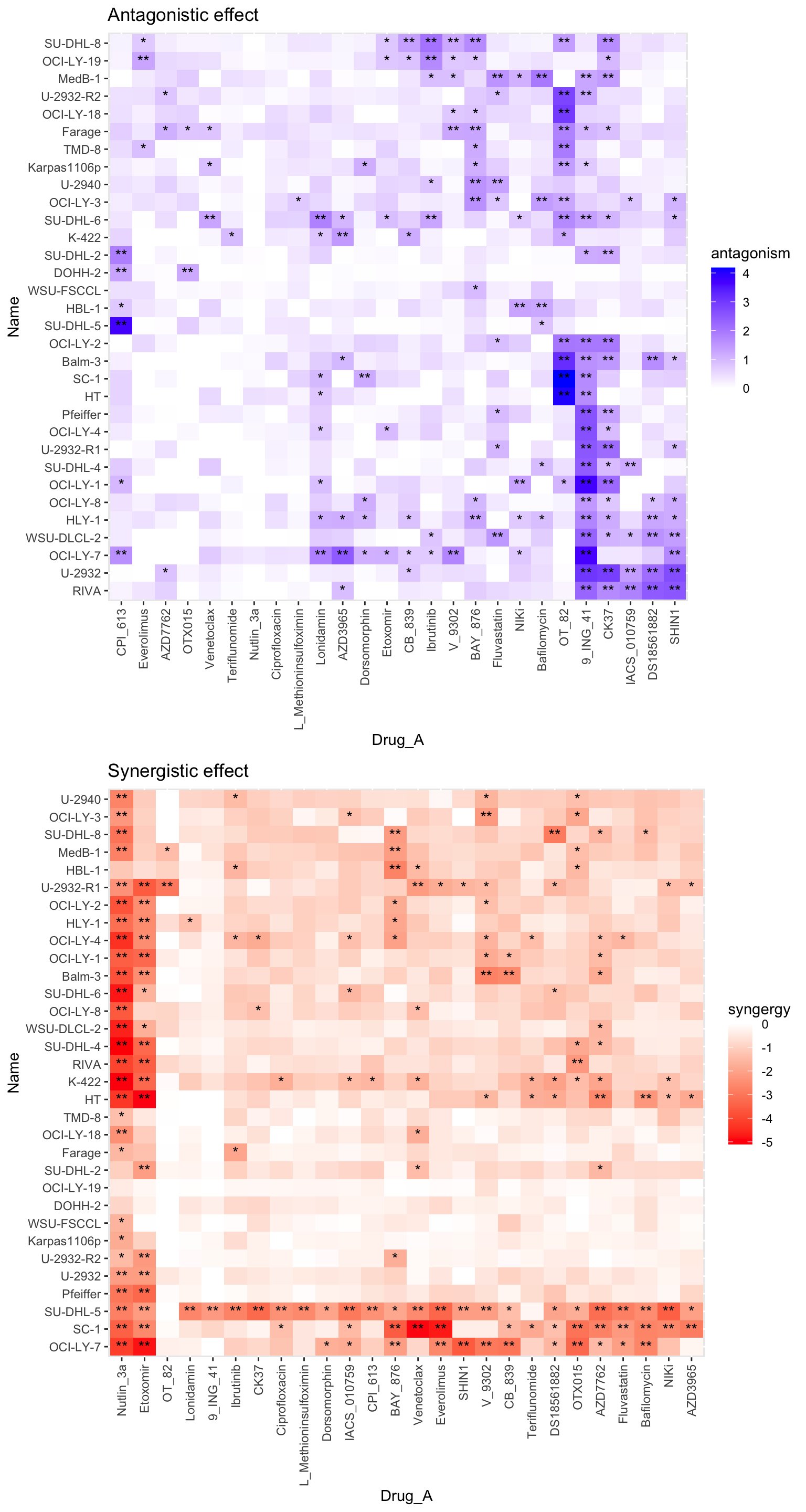 Top 10% synergistic/antagonistic effect are marked as ** and top 20% are
marked as *
Top 10% synergistic/antagonistic effect are marked as ** and top 20% are
marked as *
Correlate summarised CI to the single angent response of CHP_Pola
Add single agent CHP_Pola
polaTab <- screenData %>% filter(Drug_A == "DMSO", Drug_B == "CHP_Pola",!ifEdge) %>%
group_by(Name) %>% summarise(viabPola = mean(normVal))Test for correlation
testTab <- left_join(ciTabSum, polaTab, by = "Name") %>%
pivot_longer(c("viabPola"), names_to = "Drug_B", values_to = "viab") %>%
pivot_longer(c("syn","anta"), names_to = "effect", values_to = "score")
resTab <- group_by(testTab, Drug_A, Drug_B, effect) %>% nest() %>%
mutate(m=map(data, ~cor.test(~score+viab,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Drug_A, Drug_B, estimate, p.value, effect) %>%
ungroup() %>% group_by(effect) %>%
mutate(p.adj = p.adjust(p.value, method = "BH")) %>%
arrange(p.value)Correlation with synergistic
resTab.syn <- filter(resTab, p.adj <0.1, effect == "syn")
pList <- lapply(seq(nrow(resTab.syn)), function(i) {
rec <- resTab.syn[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Drug_B==rec$Drug_B, effect == rec$effect)
ggplot(plotTab, aes(x=viab, y=score, label = Name)) +
geom_point(aes(col=Name)) +
geom_smooth(method = "lm",se=FALSE) +
ggrepel::geom_text_repel() +
theme(legend.position = "none") +
xlab(rec$Drug_B) + ylab("average CI") +
ggtitle(sprintf("%s\n(p=%s, coef=%s)",rec$Drug_A, formatC(rec$p.value, digits = 2),formatC(rec$estimate, digits = 1)))
})
cowplot::plot_grid(plotlist=pList)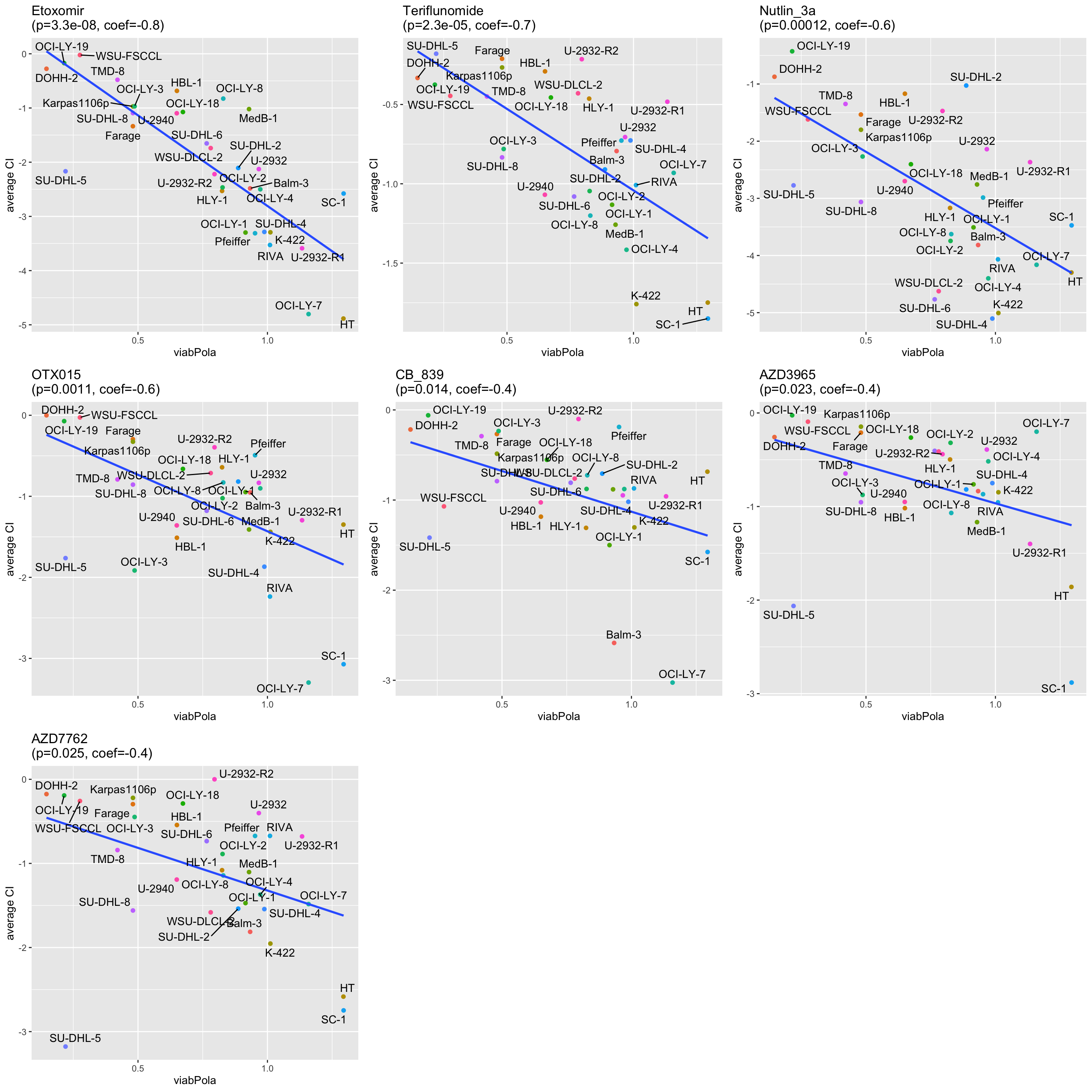
Correlation with antagonistic
resTab.anta <- filter(resTab, p.adj <0.1, effect == "anta")
pList <- lapply(seq(nrow(resTab.anta)), function(i) {
rec <- resTab.anta[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Drug_B==rec$Drug_B, effect == rec$effect)
ggplot(plotTab, aes(x=viab, y=score, label = Name)) +
geom_point(aes(col=Name)) +
geom_smooth(method = "lm",se=FALSE) +
ggrepel::geom_text_repel() +
theme(legend.position = "none") +
xlab(rec$Drug_B) + ylab("average CI") +
ggtitle(sprintf("%s\n(p=%s, coef=%s)",rec$Drug_A, formatC(rec$p.value, digits = 2),formatC(rec$estimate, digits = 1)))
})
cowplot::plot_grid(plotlist=pList)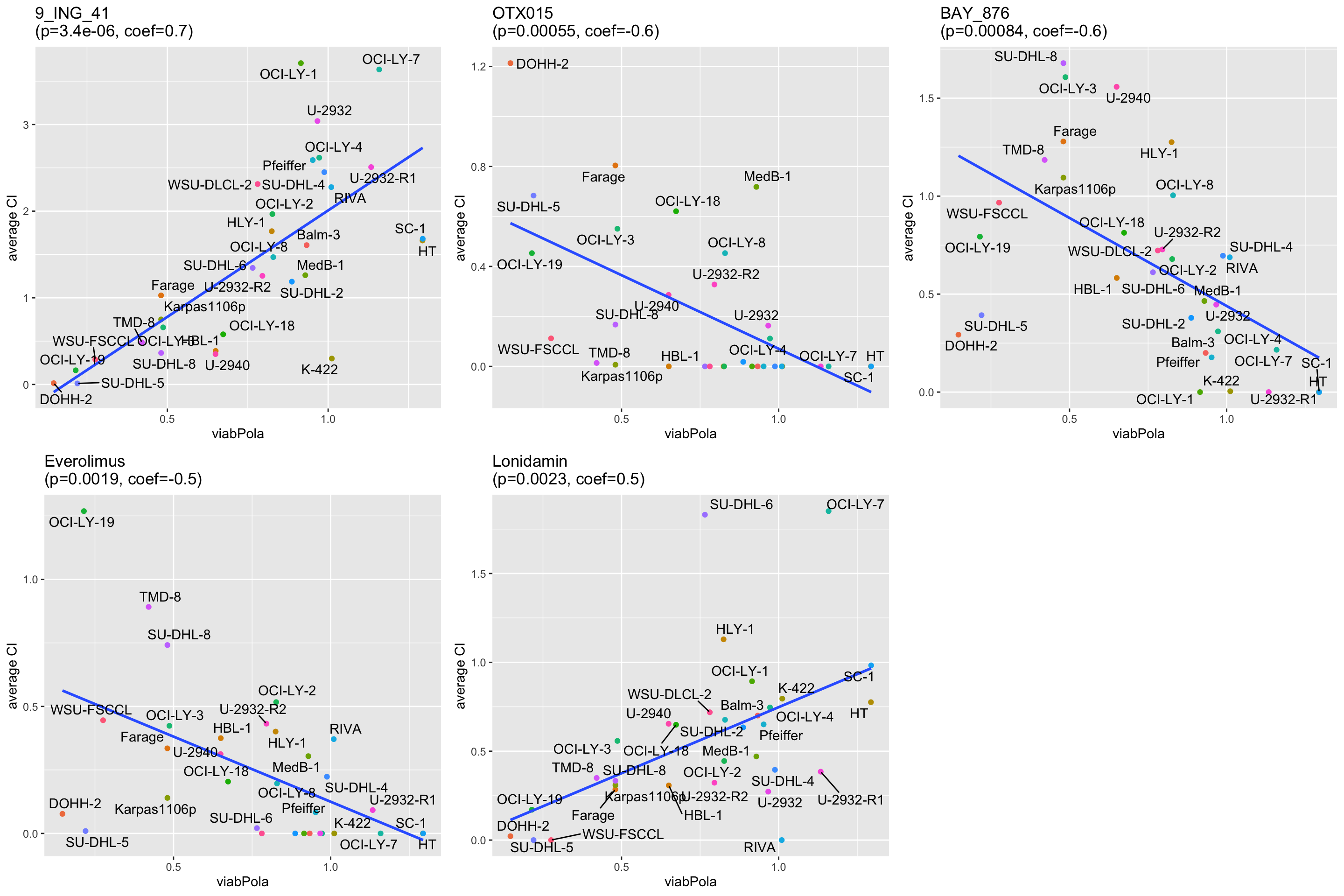
Plot combination curve for each drug, drug are ranked based on the sigificance correlation, cell lines are ranked based on CI
drugs <- unique(resTab$Drug_A)
pList <- lapply(drugs, function(eachDrug) {
ciRank <- filter(ciTabSum, Drug_A==eachDrug) %>%
arrange(syn)
plotTab <- filter(synTab, Drug_A == eachDrug) %>%
select(Name, Drug_A, Drug_A.ConcStep, viabA, viabB, viabExp, viabObs) %>%
pivot_longer(-c("Name","Drug_A","Drug_A.ConcStep"), names_to = "type", values_to = "viab") %>%
mutate(Name = factor(Name, levels = ciRank$Name)) %>%
mutate(lineType = ifelse(type %in% c("viabA","viabB"), "single","combine"))
ggplot(plotTab, aes(x=Drug_A.ConcStep, y=viab, group=type)) +
geom_line(aes(col = type, linetype = lineType)) +
facet_wrap(~Name) +
scale_linetype_manual(values = c(combine = "solid", `single` = "dotted"), name = "combination") +
scale_color_manual(values =c(viabA = "orange", viabB="darkgreen",
viabExp = "red", viabObs="blue"),
labels=c("drug_A only ","drug_B only","expected effect","observed effect"),
name = "treatment") +
ggtitle(eachDrug) +
ylab("Viability") + xlab("Concentration step")
})
jyluMisc::makepdf(pList, "../docs/combo_effect.pdf",nrow = 1, ncol = 1, height = 10, width = 10)Test for synergistic effect related to genomics
Preprocess genomics
load("../data/SVs_filtered.RData")
mutTab <- svTab %>% filter(Name %in% synTab$Name) %>%
group_by(Name, Gene) %>% summarise(n = length(Name)) %>%
arrange(desc(n))
#Get mutations occured at least in three cell lines
geneCount <- group_by(mutTab, Gene) %>% summarise(n=length(Name)) %>%
filter(n>=3) %>% arrange(desc(n))
mutTab <- filter(mutTab, Gene %in% geneCount$Gene) %>%
mutate(status =1) %>% select(Name, Gene, status) %>%
pivot_wider(names_from = "Gene", values_from = "status") %>%
mutate_all(replace_na,0) %>%
pivot_longer(-Name, names_to = "Gene", values_to = "status")T-test
testTab <- ciTabSum %>% full_join(mutTab, by = "Name") %>%
pivot_longer(c("syn","anta"), names_to = "effect", values_to = "CI") %>%
filter(!is.na(Gene),!is.na(CI), effect =="syn")
resTab <- group_by(testTab, Drug_A, Gene, effect) %>% nest() %>%
mutate(m = map(data, ~t.test(CI ~ status,., var.equal=TRUE))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Drug_A, Gene, effect, estimate, p.value) %>%
arrange(p.value) %>% ungroup() %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))
resTab.sig <- filter(resTab, p.value <= 0.05)Boxplots for associations passed p-value <= 0.05
pList <- lapply(seq(nrow(resTab.sig)), function(i) {
rec <- resTab.sig[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Gene == rec$Gene, effect == rec$effect) %>%
mutate(status = factor(status))
ggplot(plotTab, aes(x=status, y=CI)) +
geom_boxplot(aes(fill = status)) +
geom_point() +
ggtitle(sprintf("%s ~ %s (p=%s)",rec$Drug_A, rec$Gene, formatC(rec$p.value, digits = 2))) +
xlab(rec$Gene) +
theme_bw() +
theme(legend.position = "none")
})
cowplot::plot_grid(plotlist = pList, ncol=4)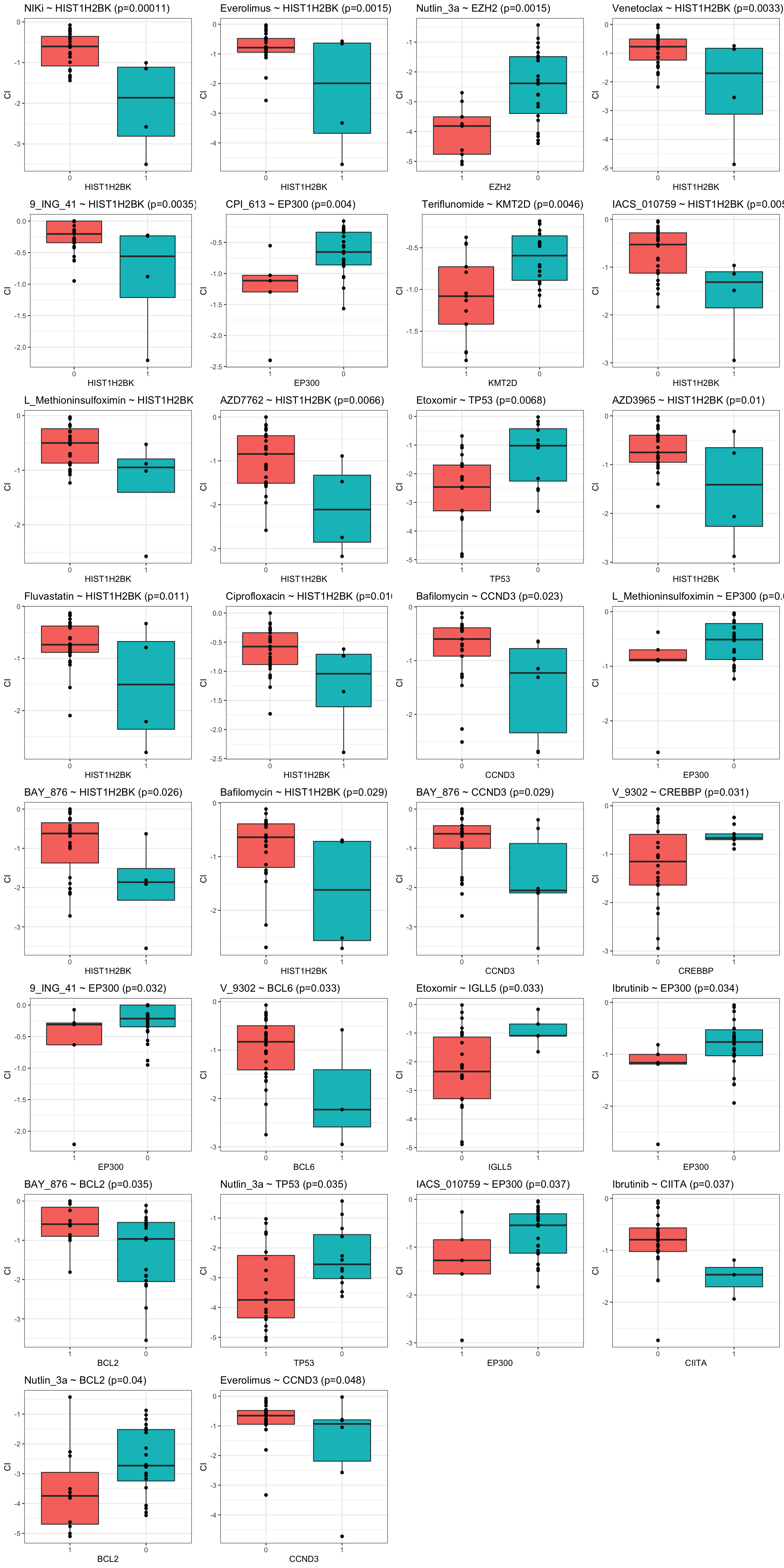
Heatmap
library(pheatmap)
ciTab <- testTab %>%
filter(Gene %in% resTab.sig$Gene, Drug_A %in% resTab.sig$Drug_A)
ciMat <- distinct(ciTab, Name, Drug_A, CI) %>%
pivot_wider(names_from = "Name", values_from = "CI") %>%
column_to_rownames("Drug_A") %>% as.matrix()
colAnno <- ciTab %>% ungroup() %>% distinct(Name, Gene, status) %>%
mutate(status = as.character(status)) %>%
pivot_wider(names_from = "Gene", values_from = "status") %>%
column_to_rownames("Name")
annoColor <- lapply(colnames(colAnno), function(x) {c("1"="black","0"="white")})
names(annoColor) <- colnames(colAnno)
pheatmap(ciMat, annotation_col = colAnno, annotation_colors = annoColor)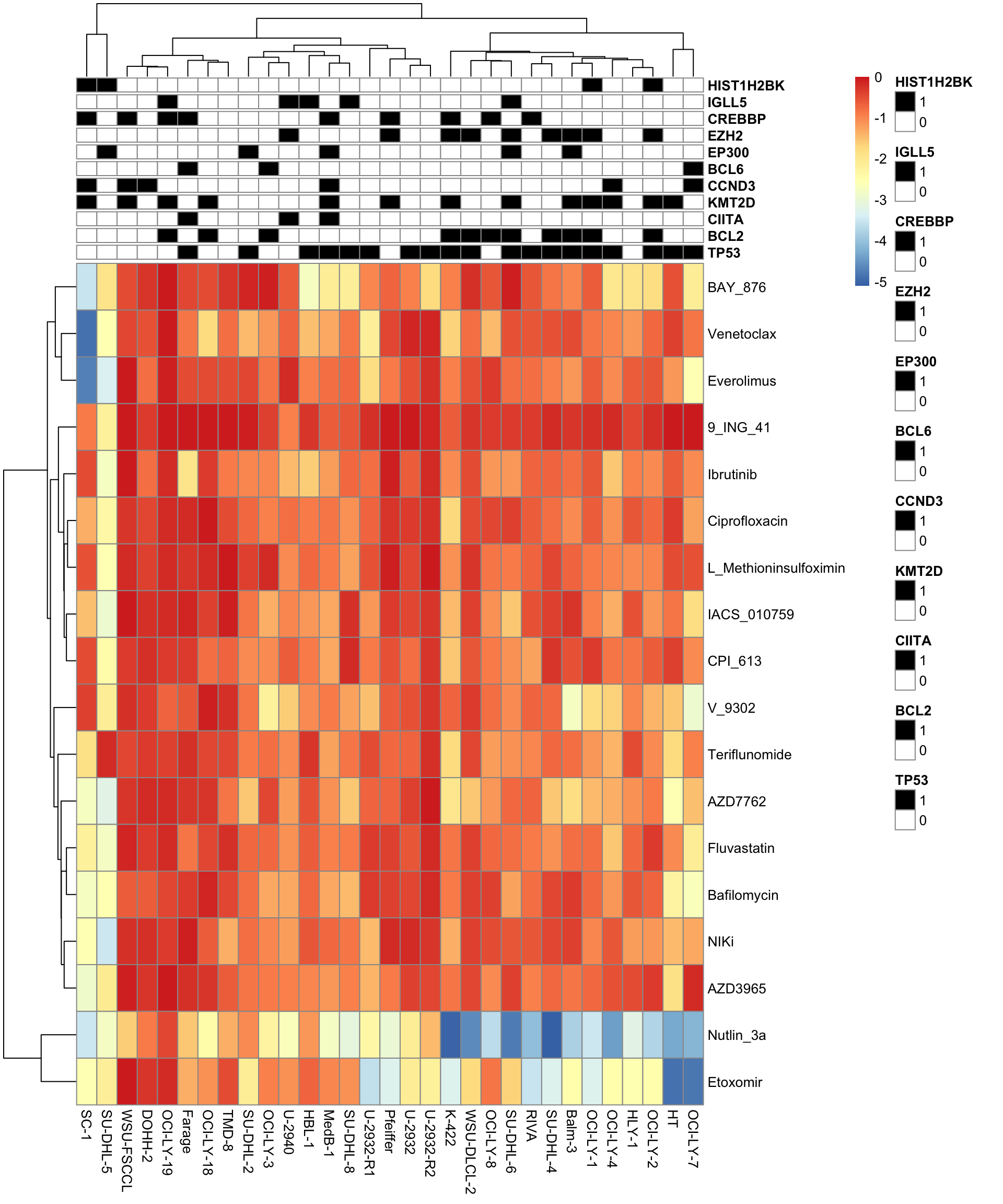
Does Etoxomir response/synergy associate with the expression of HSD17B4 or CPT1A?
Prepare Etoxomir response table
nonDLBCL <- c("Farage", "U-2940", "MedB-1", "WSU-FSCCL", "SC-1", "Karpas-1106p")
etoTabCom <- ciTabSum %>% ungroup() %>%
filter(Drug_A == "Etoxomir") %>% select(Name, syn)
etoTabSingle <- drugATab %>% filter(Drug_A=="Etoxomir") %>%
group_by(Name) %>% summarise(viab = mean(viabA))
etoTab <- left_join(etoTabCom, etoTabSingle) %>%
pivot_longer(-Name, names_to = "type", values_to = "value") %>%
filter(! Name %in% nonDLBCL ) #remove non-DLBCL cell linesTP53 mutational status (fixed)
tp53MutTab <- mutTab %>% filter(Gene == "TP53") %>%
mutate(status = ifelse(Name %in% c("Pfeiffer", "OCI-LY-8"),1,status)) %>%
mutate(TP53 = ifelse(status ==1, "Mut", "WT")) %>%
select(Name, TP53)
etoTab <- left_join(etoTab, tp53MutTab)RNAseq expression
Preprocessing
library(matrixStats)
load("../data/DepMap_GEXwide.RData")
exprMat <- t(DepMap_GEXwide)
exprMat <- exprMat[,colnames(exprMat) %in% etoTab$Name]
# Remove low count genes
#exprMat <- exprMat[rowMedians(exprMat) >0,]
#vstObj <- vsn::vsnMatrix(exprMat)
#exprMat <- vsn::predict(vstObj, exprMat)Select two interested genes
exprTab <- exprMat[c("HSD17B4","CPT1A"),] %>%
as_tibble(rownames = "Gene") %>%
pivot_longer(-Gene,names_to = "Name",values_to = "expr")Correlation test
testTab <- left_join(exprTab, etoTab, by = "Name")
resTab <- group_by(testTab, Gene, type) %>% nest() %>%
mutate(m=map(data,~cor.test(~expr+value,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Gene, type, p.value, estimate)
resTab# A tibble: 4 × 4
# Groups: Gene, type [4]
Gene type p.value estimate
<chr> <chr> <dbl> <dbl>
1 HSD17B4 syn 0.102 -0.474
2 HSD17B4 viab 0.840 -0.0621
3 CPT1A syn 0.00311 -0.751
4 CPT1A viab 0.413 -0.249 “Syn” indicates synergy (CI values), “viab” indicates single agent response
Correlation plot
ggplot(testTab, aes(x=expr, y = value)) +
ggrepel::geom_text_repel(aes(label = Name)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
facet_wrap(~Gene + type, scale = "free")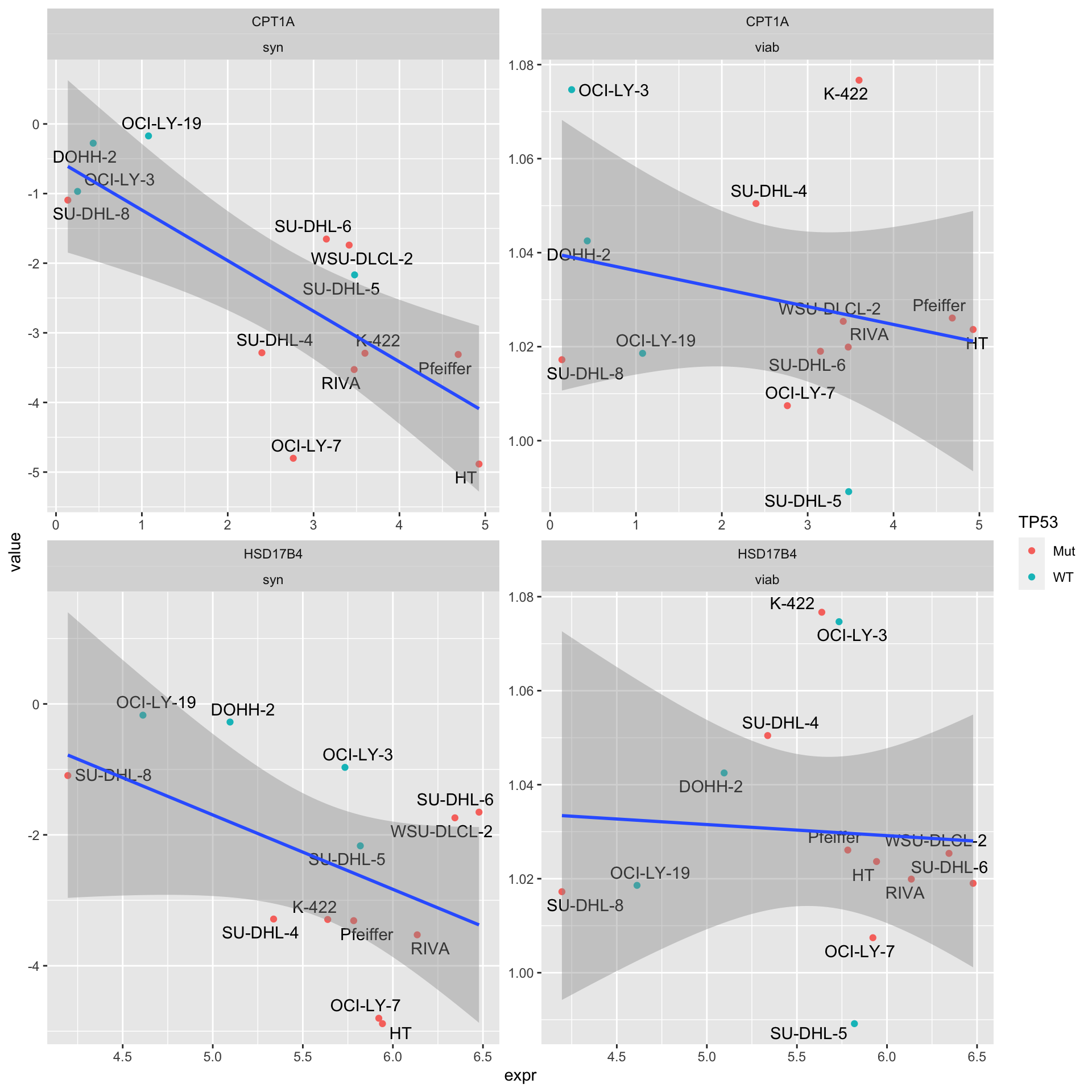
Proteom expression (SMART-CARE)
Preprocessing
library(SummarizedExperiment)
protData <- readRDS("../data/SC005_SummarizedExperiment_proteomics.RDS")
#select baseline samples
protData <- protData[rowData(protData)$Gene_name %in% c("HSD17B4","CPT1A"), protData$cell.line %in% etoTab$Name & protData$condition == "U"]
exprMat <- assay(protData)
rownames(exprMat) <- rowData(protData)$Gene_nameSelect two interested genes
protTab <- exprMat[c("HSD17B4","CPT1A"),] %>%
as_tibble(rownames = "Gene") %>%
pivot_longer(-Gene,names_to = "smp",values_to = "expr") %>%
mutate(Name = protData[,smp]$cell.line) %>%
group_by(Gene,Name) %>%
summarise(expr = mean(expr, na.rm=TRUE)) %>% ungroup() %>%
filter(!is.na(expr))Correlation test
testTab <- left_join(protTab, etoTab, by = "Name")
resTab <- group_by(testTab, Gene, type) %>% nest() %>%
mutate(m=map(data,~cor.test(~expr+value,., use= "pairwise.complete.obs"))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Gene, type, p.value, estimate)
resTab# A tibble: 4 × 4
# Groups: Gene, type [4]
Gene type p.value estimate
<chr> <chr> <dbl> <dbl>
1 CPT1A syn 0.678 -0.150
2 CPT1A viab 0.999 0.000656
3 HSD17B4 syn 0.0235 -0.645
4 HSD17B4 viab 0.471 0.230 “Syn” indicates synergy (CI values), “viab” indicates single agent response
Correlation plot
ggplot(testTab, aes(x=expr, y = value)) +
ggrepel::geom_text_repel(aes(label = Name)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
facet_wrap(~Gene + type, scale = "free")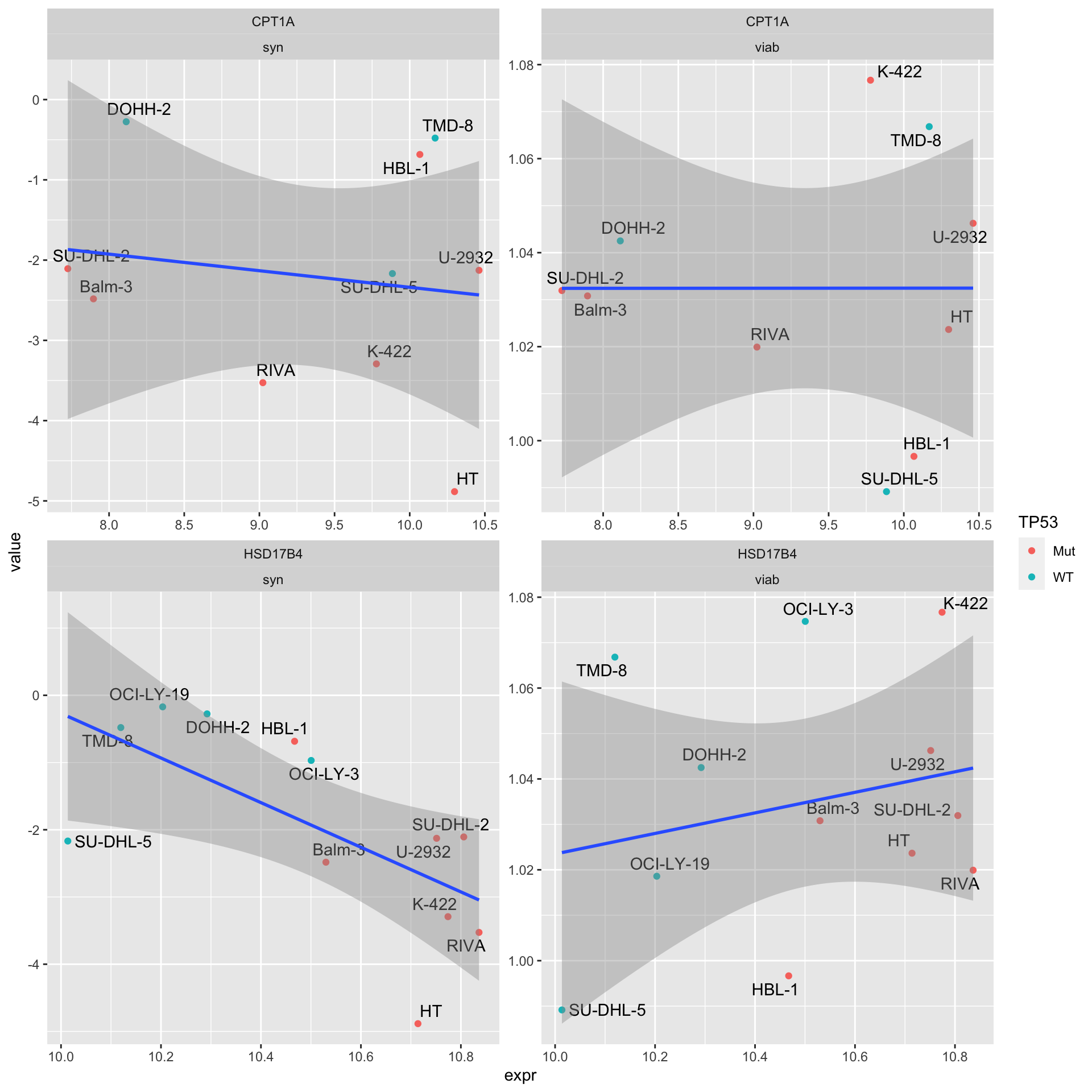
Proteom expression (EMBL)
Preprocessing
load("../data/ProtWide.RData")
ProtWide <- ProtWide[,colnames(ProtWide) %in% etoTab$Name]
exprMat <- ProtWideSelect two interested genes
protTab1 <- exprMat[c("HSD17B4","CPT1A"),] %>%
as_tibble(rownames = "Gene") %>%
pivot_longer(-Gene,names_to = "Name",values_to = "expr")Correlation test
testTab <- left_join(protTab1, etoTab, by = "Name")
resTab <- group_by(testTab, Gene, type) %>% nest() %>%
mutate(m=map(data,~cor.test(~expr+value,., use= "pairwise.complete.obs"))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Gene, type, p.value, estimate)
resTab# A tibble: 4 × 4
# Groups: Gene, type [4]
Gene type p.value estimate
<chr> <chr> <dbl> <dbl>
1 HSD17B4 syn 0.0617 -0.372
2 HSD17B4 viab 0.561 -0.120
3 CPT1A syn 0.105 -0.326
4 CPT1A viab 0.942 -0.0150“Syn” indicates synergy (CI values), “viab” indicates single agent response
Correlation plot
ggplot(testTab, aes(x=expr, y = value)) +
ggrepel::geom_text_repel(aes(label = Name)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
facet_wrap(~Gene + type, scale = "free")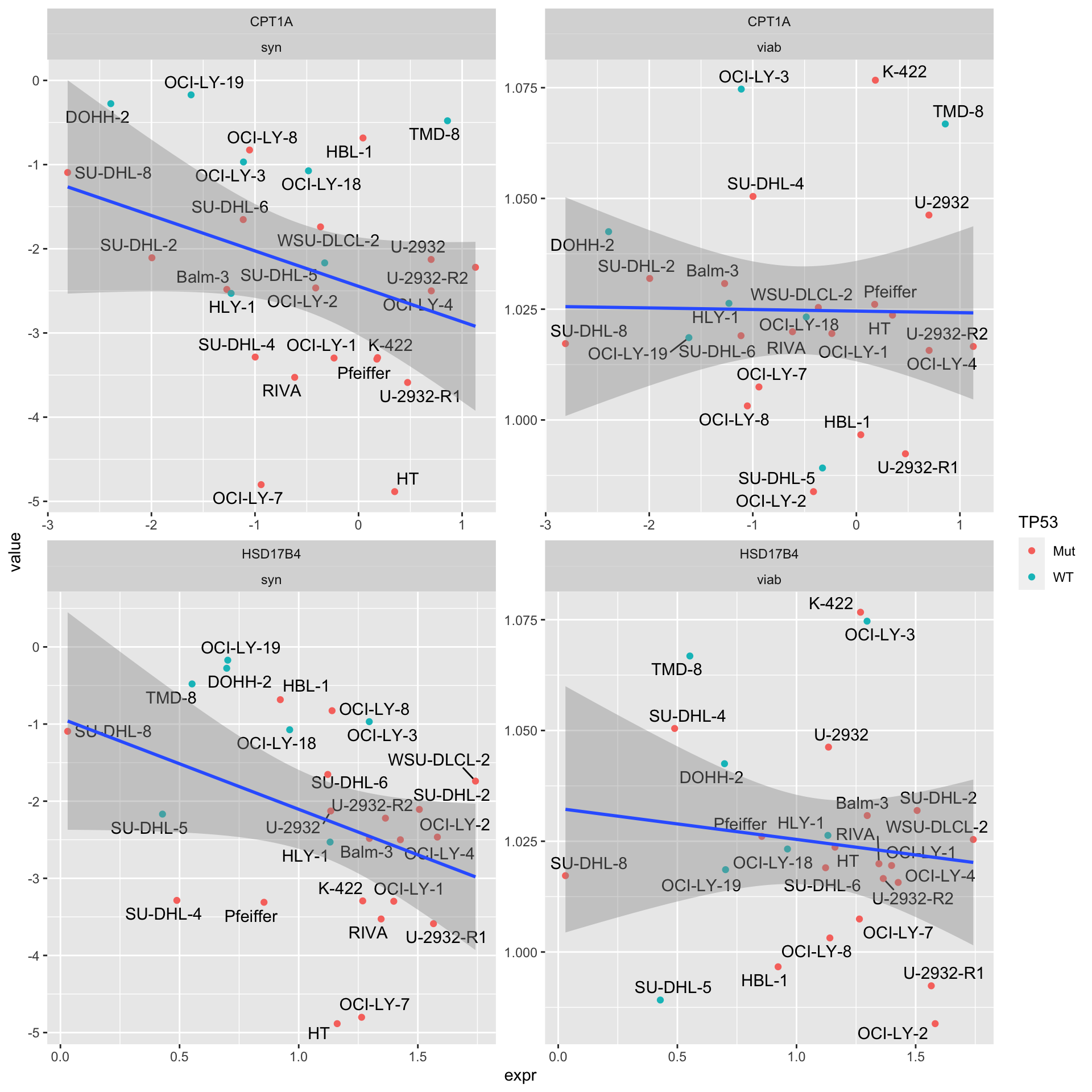
Does Etoxomir response/synergy associate with the abundance of any metabolites?
Preprocessing
metaData <- readRDS("../data/SC005_SummarizedExperiment_metabolomics.RDS")
metaData <- metaData[metaData$condition == "U",metaData$cell.line %in% etoTab$Name]
metaMat <- assay(metaData)
metaMatNorm <- PhosR::medianScaling(metaMat, scale = FALSE)
#vsnFit <- vsn::vsnMatrix(metaMat)
#metaMatNorm <- metaMat
#boxplot(metaMatNorm)
metaNorm <- metaData
assay(metaNorm) <- metaMatNorm
assayNames(metaNorm) <- "norm"
exprMat <- assay(metaNorm)Select two interested genes
metaTab <- exprMat %>%
as_tibble(rownames = "Metabolite") %>%
pivot_longer(-Metabolite,names_to = "smp",values_to = "expr") %>%
mutate(Name = metaNorm[,smp]$cell.line) %>%
group_by(Metabolite,Name) %>%
summarise(expr = mean(expr, na.rm=TRUE)) %>% ungroup() %>%
filter(!is.na(expr))Correlation test
testTab <- left_join(metaTab, etoTab, by = "Name")
resTab <- group_by(testTab, Metabolite, type) %>% nest() %>%
mutate(m=map(data,~cor.test(~expr+value,., use= "pairwise.complete.obs"))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Metabolite, type, p.value, estimate) %>%
arrange(p.value)
resTab# A tibble: 286 × 4
# Groups: Metabolite, type [286]
Metabolite type p.value estimate
<chr> <chr> <dbl> <dbl>
1 Hex2Cer(d18:1/24:0) syn 0.00107 0.821
2 HexCer(d18:2/16:0) syn 0.0184 -0.664
3 DG-O(16:0_18:1) syn 0.0204 -0.656
4 Cer(d18:2/24:1) syn 0.0212 -0.653
5 Hex2Cer(d18:1/26:1) syn 0.0229 0.647
6 Betaine syn 0.0244 -0.642
7 HexCer(d18:2/24:0) viab 0.0247 -0.641
8 Hex2Cer(d18:1/24:1) syn 0.0280 0.630
9 HexCer(d18:2/24:0) syn 0.0285 -0.629
10 PC ae C34:1 syn 0.0313 -0.620
# … with 276 more rows
# ℹ Use `print(n = ...)` to see more rows“Syn” indicates synergy (CI values), “viab” indicates single agent response
Correlation plot
ggplot(filter(testTab, Metabolite %in% resTab[1:9,]$Metabolite), aes(x=expr, y = value)) +
ggrepel::geom_text_repel(aes(label = Name)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
facet_wrap(~Metabolite + type, scale = "free")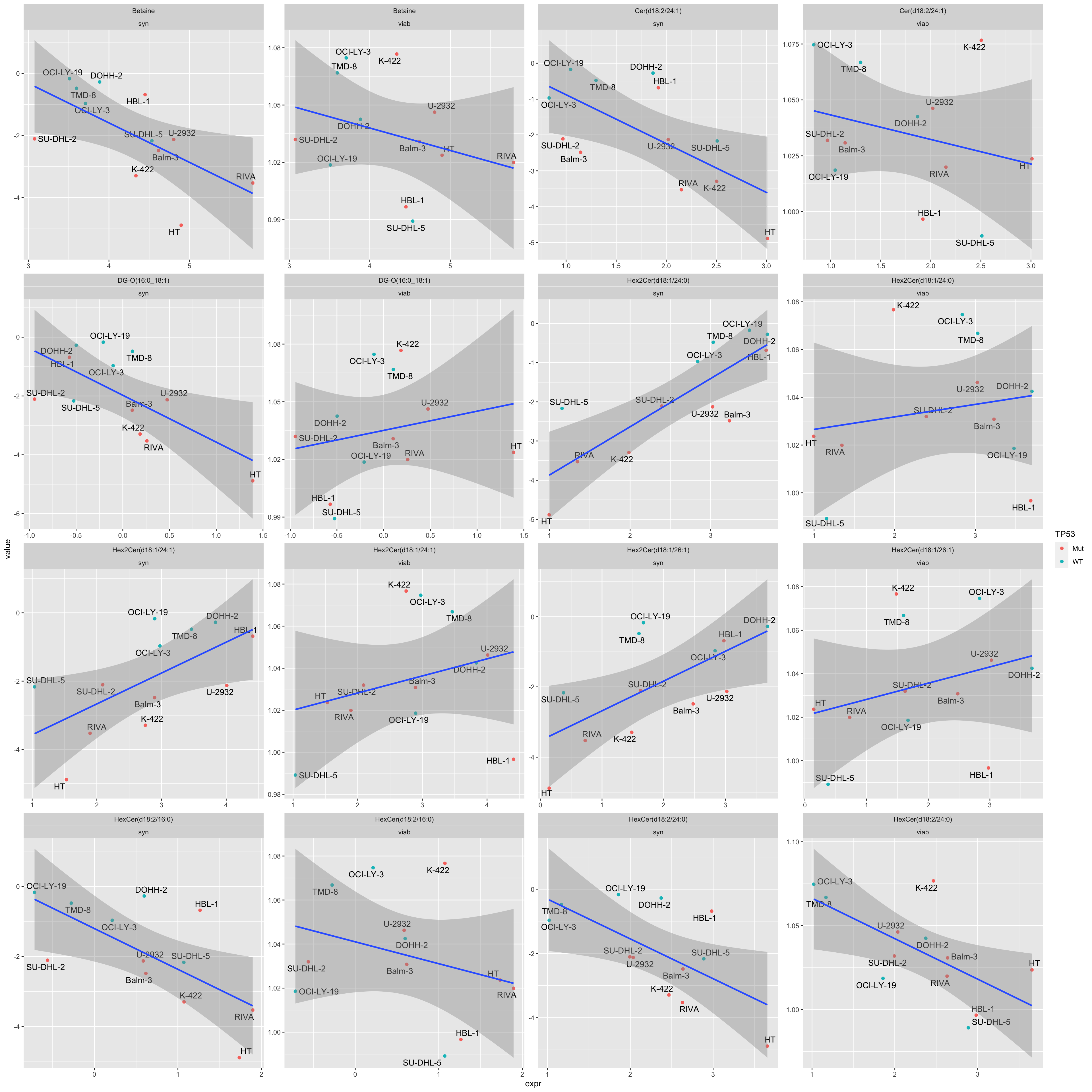
Correlation gene/protein expression of HSD17B4 and CPT1A with metabolites
exprTabAll <- bind_rows(mutate(exprTab, set = "RNA"),
mutate(protTab, set = "Protein_SMART"),
mutate(protTab1, set = "Protein_EMBL"))
testTab <- full_join(exprTabAll, dplyr::rename(metaTab, abundance = expr), by = "Name") %>%
filter(!is.na(expr), !is.na(abundance)) %>%
left_join(tp53MutTab)resTab <- group_by(testTab, Gene, set, Metabolite) %>% nest() %>%
mutate(m=map(data, ~cor.test(~expr+abundance,.))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Gene, set, Metabolite, estimate, p.value) %>%
arrange(p.value) %>% ungroup() %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))Result table (FDR < 0.1)
resTab.sig <- filter(resTab, p.adj <= 0.1)
resTab.sig %>% mutate_if(is.numeric, formatC, digits=2) %>%
DT::datatable()Correlation plots of significant associations
pList <- lapply(seq(9), function(i) {
rec <- resTab.sig[i,]
plotTab <- testTab %>% filter(Gene == rec$Gene, set == rec$set, Metabolite == rec$Metabolite)
ggplot(plotTab, aes(x=expr, y=abundance)) +
geom_point(aes(col = TP53)) +
geom_smooth(method = "lm") +
ggrepel::geom_text_repel(aes(label = Name)) +
ggtitle(sprintf("%s ~ %s\n(P=%s, %s)", rec$Metabolite, rec$Gene, formatC(rec$p.value, digits = 2), rec$set)) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList, ncol=3)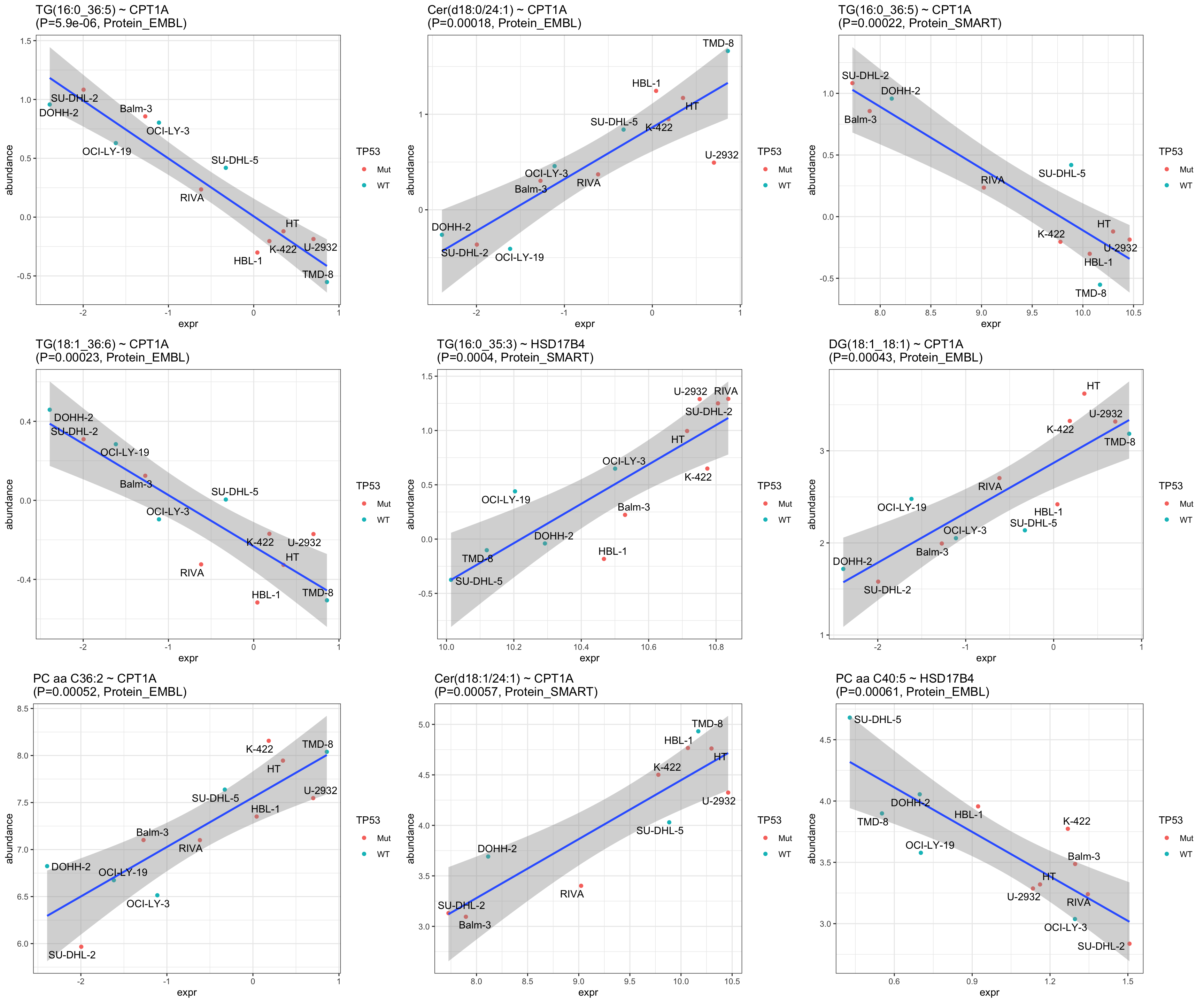
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] SummarizedExperiment_1.26.1 Biobase_2.56.0
[3] GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[5] IRanges_2.30.0 S4Vectors_0.34.0
[7] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[9] matrixStats_0.62.0 pheatmap_1.0.12
[11] gridExtra_2.3 forcats_0.5.1
[13] stringr_1.4.0 dplyr_1.0.9
[15] purrr_0.3.4 readr_2.1.2
[17] tidyr_1.2.0 tibble_3.1.8
[19] ggplot2_3.3.6 tidyverse_1.3.2
loaded via a namespace (and not attached):
[1] utf8_1.2.2 shinydashboard_0.7.2 tidyselect_1.1.2
[4] htmlwidgets_1.5.4 grid_4.2.0 BiocParallel_1.30.3
[7] maxstat_0.7-25 munsell_0.5.0 preprocessCore_1.58.0
[10] codetools_0.2-18 DT_0.23 withr_2.5.0
[13] colorspace_2.0-3 highr_0.9 knitr_1.39
[16] rstudioapi_0.13 ggsignif_0.6.3 labeling_0.4.2
[19] git2r_0.30.1 slam_0.1-50 GenomeInfoDbData_1.2.8
[22] KMsurv_0.1-5 farver_2.1.1 rprojroot_2.0.3
[25] coda_0.19-4 vctrs_0.4.1 generics_0.1.3
[28] TH.data_1.1-1 xfun_0.31 sets_1.0-21
[31] R6_2.5.1 bitops_1.0-7 cachem_1.0.6
[34] reshape_0.8.9 fgsea_1.22.0 DelayedArray_0.22.0
[37] assertthat_0.2.1 promises_1.2.0.1 scales_1.2.0
[40] multcomp_1.4-19 googlesheets4_1.0.0 gtable_0.3.0
[43] sandwich_3.0-2 workflowr_1.7.0 rlang_1.0.4
[46] GlobalOptions_0.1.2 splines_4.2.0 rstatix_0.7.0
[49] gargle_1.2.0 broom_1.0.0 reshape2_1.4.4
[52] yaml_2.3.5 abind_1.4-5 modelr_0.1.8
[55] crosstalk_1.2.0 backports_1.4.1 httpuv_1.6.5
[58] tools_4.2.0 relations_0.6-12 statnet.common_4.6.0
[61] ellipsis_0.3.2 gplots_3.1.3 jquerylib_0.1.4
[64] RColorBrewer_1.1-3 ggdendro_0.1.23 proxy_0.4-27
[67] Rcpp_1.0.9 plyr_1.8.7 visNetwork_2.1.0
[70] zlibbioc_1.42.0 RCurl_1.98-1.7 ggpubr_0.4.0
[73] viridis_0.6.2 cowplot_1.1.1 zoo_1.8-10
[76] haven_2.5.0 ggrepel_0.9.1 cluster_2.1.3
[79] exactRankTests_0.8-35 fs_1.5.2 magrittr_2.0.3
[82] data.table_1.14.2 PhosR_1.6.0 circlize_0.4.15
[85] reprex_2.0.1 survminer_0.4.9 pcaMethods_1.88.0
[88] googledrive_2.0.0 mvtnorm_1.1-3 hms_1.1.1
[91] shinyjs_2.1.0 mime_0.12 evaluate_0.15
[94] xtable_1.8-4 readxl_1.4.0 shape_1.4.6
[97] compiler_4.2.0 KernSmooth_2.23-20 crayon_1.5.1
[100] htmltools_0.5.3 mgcv_1.8-40 later_1.3.0
[103] tzdb_0.3.0 lubridate_1.8.0 DBI_1.1.3
[106] dbplyr_2.2.1 MASS_7.3-58 jyluMisc_0.1.5
[109] Matrix_1.4-1 car_3.1-0 cli_3.3.0
[112] marray_1.74.0 parallel_4.2.0 igraph_1.3.4
[115] pkgconfig_2.0.3 km.ci_0.5-6 piano_2.12.0
[118] xml2_1.3.3 bslib_0.4.0 ruv_0.9.7.1
[121] XVector_0.36.0 drc_3.0-1 rvest_1.0.2
[124] digest_0.6.29 rmarkdown_2.14 cellranger_1.1.0
[127] fastmatch_1.1-3 survMisc_0.5.6 dendextend_1.16.0
[130] shiny_1.7.2 gtools_3.9.3 lifecycle_1.0.1
[133] nlme_3.1-158 jsonlite_1.8.0 carData_3.0-5
[136] network_1.17.2 viridisLite_0.4.0 limma_3.52.2
[139] fansi_1.0.3 pillar_1.8.0 lattice_0.20-45
[142] GGally_2.1.2 fastmap_1.1.0 httr_1.4.3
[145] plotrix_3.8-2 survival_3.3-1 glue_1.6.2
[148] class_7.3-20 stringi_1.7.8 sass_0.4.2
[151] caTools_1.18.2 e1071_1.7-11