Analysis of combinatorial effect using simple bliss model
Junyan Lu
2022-04-25
Last updated: 2023-06-16
Checks: 5 1
Knit directory: combiDLBCL/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20220425) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the
results is critical for reproducibility. To start using Git, open the
Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full
reproducibility benefits of using workflowr, please see
?wflow_start.
Load libraries and datasets
Calculate CI for the combination of bases and combi drugs
screenSub <- screenData %>%
mutate(Name = cellLine, normVal = normVal.cor) %>%
filter(!ifRemove)Get combination effect
comTab <- filter(screenSub, Drug_A !="DMSO", Drug_B != "DMSO") %>%
select(Name, Drug_A, Drug_A.Conc, Drug_B, Drug_B.Conc, normVal) %>%
dplyr::rename(viabObs = normVal) %>%
distinct(Name, Drug_A, Drug_A.Conc, Drug_B, Drug_B.Conc, .keep_all = TRUE)
drugATab <- filter(screenSub, Drug_A != "DMSO", Drug_B == "DMSO") %>%
select(Name, Drug_A, Drug_A.Conc, normVal, Drug_A.ConcStep) %>%
dplyr::rename(viabA = normVal) %>%
distinct(Name, Drug_A, Drug_A.Conc, .keep_all = TRUE)
drugBTab <- filter(screenSub, Drug_A == "DMSO", Drug_B != "DMSO") %>%
select(Name, Drug_B, Drug_B.Conc, normVal, Drug_B.ConcStep) %>%
dplyr::rename(viabB = normVal) %>%
distinct(Name,Drug_B, Drug_B.Conc, .keep_all = TRUE)synTab <- comTab %>% left_join(drugATab, by =c("Name","Drug_A","Drug_A.Conc")) %>%
left_join(drugBTab, by = c("Name","Drug_B","Drug_B.Conc")) %>%
mutate(viabExp = viabA*viabB) %>%
mutate(CI = viabObs-viabExp,
logCI = log10(viabObs/viabExp))Save CI table for shiny app
save(synTab, file = "../shiny/combiDLBCL/synTab_AA2022.RData")Visualize CI for each drug and concentration
pList <- lapply(unique(synTab$Drug_A), function(drugA) {
plotTab <- filter(synTab, Drug_A == drugA)
ggplot(plotTab, aes(x=Drug_B.ConcStep, y=Drug_B, fill = CI)) +
geom_tile() +
facet_wrap(~Name, ncol=5) +
scale_fill_gradient2(low ="red",high="blue",mid="white") +
ggtitle(drugA)
})
names(pList) <- unique(synTab$Drug_A)
jyluMisc::makepdf(pList, "../docs/CI_heatmap_AAscreen2023.pdf", height = 30, width = 10, ncol = 1, nrow = 1)Summarise CI
Calculate synergistic and antagonistic effect separately, using a similar way as bayesyngergy package
sumSyn <- function(viabExp, viabObs) {
tab <- tibble(viabExp=viabExp, viabObs = viabObs) %>%
mutate(syn = min(0, viabObs - viabExp),
anta = max(0, viabObs - viabExp))
return(tibble(syn = sum(tab$syn,na.rm = TRUE), anta = sum(tab$anta,na.rm = TRUE)))
}
ciTabSum <- group_by(synTab, Name, Drug_A, Drug_B) %>% nest() %>%
mutate(res = map(data, ~sumSyn(.$viabExp, .$viabObs))) %>%
unnest(res) %>% select(-data)Visualization of synergistic and antagoistic effect
plotSynScatter <- function(plotTab, labelPercent = 0.05) {
synCut <- quantile(plotTab$syn, labelPercent)
antaCut <- quantile(plotTab$anta, 1-labelPercent)
plotTabSyn <- plotTab %>% mutate(labelText = ifelse(syn < synCut, Name,""), type = "Synergistic effect") %>%
mutate(score = syn)
plotTabAnta <- plotTab %>% mutate(labelText = ifelse(anta > antaCut, Name,""), type = "Antagonistic effect") %>%
mutate(score = anta)
plotComb <- bind_rows(plotTabSyn, plotTabAnta)
p <- ggplot(plotComb, aes(x=Drug_B, y=score, label = labelText)) +
geom_point(aes(col= type), alpha=0.5) + ggrepel::geom_text_repel(max.overlaps = Inf) +
theme_bw() +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust = 0.5)) +
scale_color_manual(values = c("Synergistic effect" = "red", "Antagonistic effect" = "blue")) +
#facet_wrap(~type, ncol=2)
ggtitle(unique(plotComb$Drug_A)) + ylab("Combination Score") + xlab("") +
theme(legend.position = "bottom")
return(p)
}Scatter plot
Top 5% synergistics or antagonistic effect are labelled.
pList <- lapply(unique(ciTabSum$Drug_A), function(drugA){
plotTab <- filter(ciTabSum, Drug_A == drugA)
plotSynScatter(plotTab,0.05)
})
jyluMisc::makepdf(pList, "../docs/synegy_per_DrugA_AAscreen2023.pdf", height = 10, width = 10, ncol = 1, nrow = 1)Matrix visualization
plotSynMatrix <- function(ciTabSum, cut1 = 0.1, cut2 = 0.2) {
synCut1 <- quantile(ciTabSum$syn, cut1)
antaCut1 <- quantile(ciTabSum$anta, 1-cut1)
synCut2 <- quantile(ciTabSum$syn, cut2)
antaCut2 <- quantile(ciTabSum$anta, 1-cut2)
synOrder <- hcOrder(ciTabSum$Drug_B, ciTabSum$Name, ciTabSum$syn)
antaOrder <- hcOrder(ciTabSum$Drug_B, ciTabSum$Name, ciTabSum$anta)
plotTabSyn <- ciTabSum %>% mutate(degree = case_when(
syn <= synCut1 ~ "**",
syn <= synCut2 ~ "*",
TRUE ~ ""
)) %>% mutate(Drug_B = factor(Drug_B, levels = synOrder$row),
Name = factor(Name, levels = synOrder$col))
plotTabAnta <- ciTabSum %>% mutate(degree = case_when(
anta >= antaCut1 ~ "**",
anta >= antaCut2 ~ "*",
TRUE ~ ""
)) %>% mutate(Drug_B = factor(Drug_B, levels = antaOrder$row),
Name = factor(Name, levels = antaOrder$col))
p1 <- ggplot(plotTabSyn, aes(x=Drug_B, y=Name, label = degree)) +
geom_tile(aes(fill = syn)) +
scale_fill_gradient(low="red", high="white", name = "syngergy") +
geom_text() + ggtitle(sprintf("%s (Synergistic effect)", unique(plotTabSyn$Drug_A))) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust=0.5))
p2 <- ggplot(plotTabAnta, aes(x=Drug_B, y=Name, label = degree)) +
geom_tile(aes(fill = anta)) +
scale_fill_gradient(low="white", high="blue", name = "antagonism") +
geom_text() + ggtitle(sprintf("%s (Antagonistic effect)",unique(plotTabAnta$Drug_A))) +
theme(axis.text.x = element_text(angle = 90, hjust=1, vjust=0.5))
p <- cowplot::plot_grid(p2, p1, ncol=2)
return(p)
}pList <- lapply(unique(ciTabSum$Drug_A), function(drugA) {
plotTab <- filter(ciTabSum, Drug_A == drugA)
plotSynMatrix(plotTab, 0.01, 0.05)
})
jyluMisc::makepdf(pList, "../docs/synegyHeatmap_per_DrugA_AAscreen2023.pdf", height = 8, width = 15, ncol = 1, nrow = 1)Top 1% synergistic/antagonistic effect are marked as ** and top 5% are marked as * synegyHeatmap_per_DrugA_AAscreen2023.pdf
Dose-response plots for synergistic effect
Use the shiny app for the visualization: http://mozi.embl.de/combiDLBCL/
Test for synergistic effect related to genomics (SNVs)
Preprocess genomics
load("../data/SVs_filtered.RData")
mutTab <- svTab %>% filter(Name %in% synTab$Name) %>%
group_by(Name, Gene) %>% summarise(n = length(Name)) %>%
arrange(desc(n))
#Get mutations occured at least in three cell lines
geneCount <- group_by(mutTab, Gene) %>% summarise(n=length(Name)) %>%
filter(n>=3) %>% arrange(desc(n))
mutTab <- filter(mutTab, Gene %in% geneCount$Gene) %>%
mutate(status =1) %>% select(Name, Gene, status) %>%
pivot_wider(names_from = "Gene", values_from = "status") %>%
mutate_all(replace_na,0) %>%
pivot_longer(-Name, names_to = "Gene", values_to = "status")T-test
testTab <- ciTabSum %>% full_join(mutTab, by = "Name") %>%
pivot_longer(c("syn","anta"), names_to = "effect", values_to = "CI") %>%
filter(!is.na(Gene),!is.na(CI), effect =="syn")
resTab <- group_by(testTab, Drug_A, Drug_B, Gene, effect) %>% nest() %>%
mutate(m = map(data, ~t.test(CI ~ status,., var.equal=TRUE))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
select(Drug_A, Drug_B, Gene, effect, estimate, p.value) %>%
arrange(p.value) %>% ungroup() %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))
resTab.sig <- filter(resTab, p.adj<0.25)
resTab.sig %>% mutate_if(is.numeric, formatC, digits=1) %>% DT::datatable()Boxplots for associations passed p.adj < 0.25
pList <- lapply(seq(nrow(resTab.sig)), function(i) {
rec <- resTab.sig[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Drug_B==rec$Drug_B, Gene == rec$Gene, effect == rec$effect) %>%
mutate(status = factor(status, levels = c(1,0)))
ggplot(plotTab, aes(x=status, y=CI)) +
geom_boxplot(aes(fill = status)) +
geom_point() +
ggtitle(sprintf("%s + %s ~ %s\n(p=%s)",rec$Drug_A,rec$Drug_B, rec$Gene, formatC(rec$p.value, digits = 2))) +
xlab(rec$Gene) +
theme_bw() +
theme(legend.position = "none")
})
cowplot::plot_grid(plotlist = pList, ncol=4)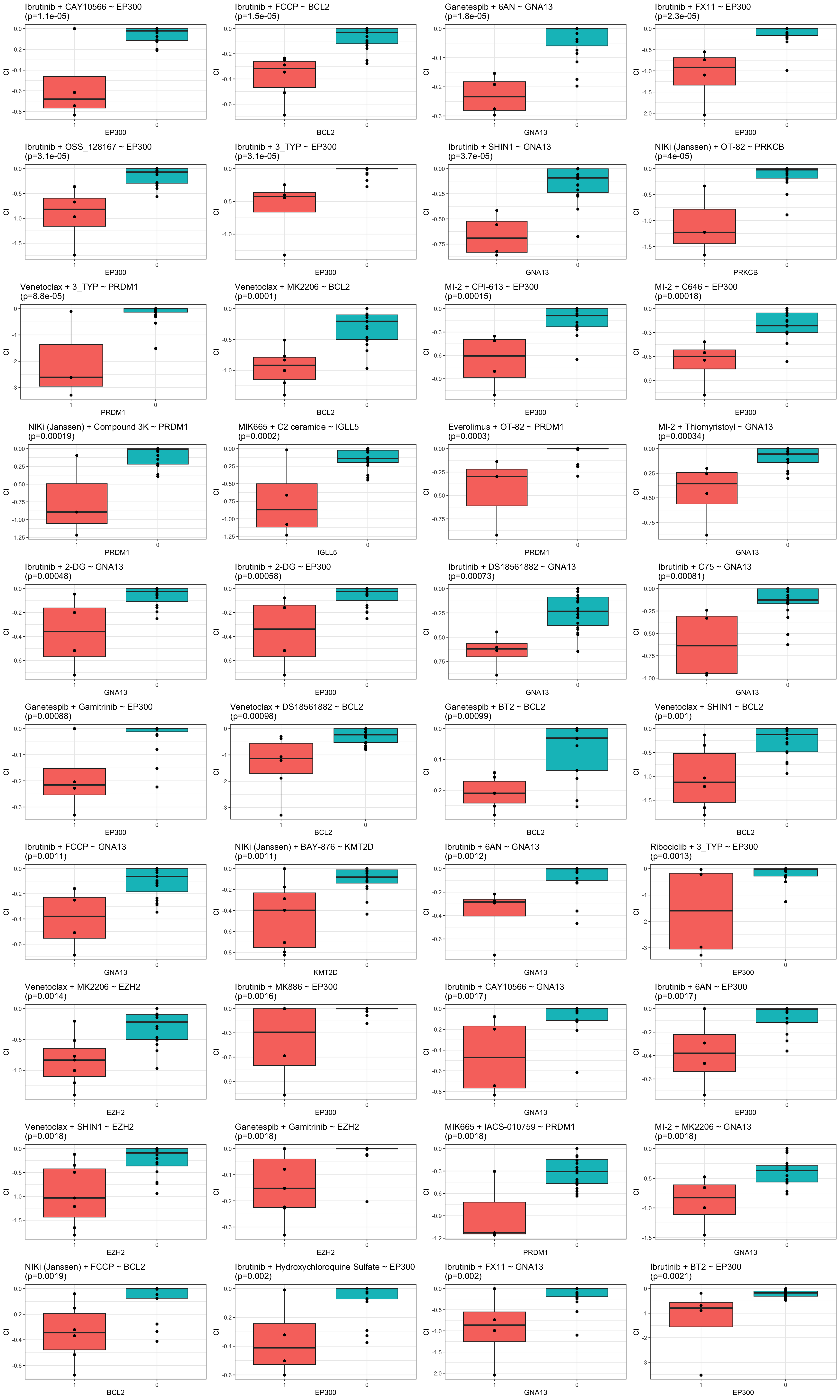
Correlate combination effect with clinical drug responses
Test per concentration
resTab.conc <- synTab %>% filter(!is.na(viabA), !is.na(CI)) %>%
group_by(Drug_A, Drug_A.Conc, Drug_A.ConcStep,
Drug_B, Drug_B.ConcStep) %>% nest() %>%
mutate(m = map(data, ~cor.test(~CI+viabA,.,method = "spearman"))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>% ungroup() %>%
arrange(p.value) %>%
group_by(Drug_A) %>%
mutate(p.adj = p.adjust(p.value, method = "BH"))P-value histogram per clinical drug
ggplot(resTab.conc, aes(x=p.value)) +
geom_histogram(aes(fill = Drug_A), alpha=0.5, color = "black") +
facet_wrap(~Drug_A) +
theme_bw() +
theme(legend.position = "none")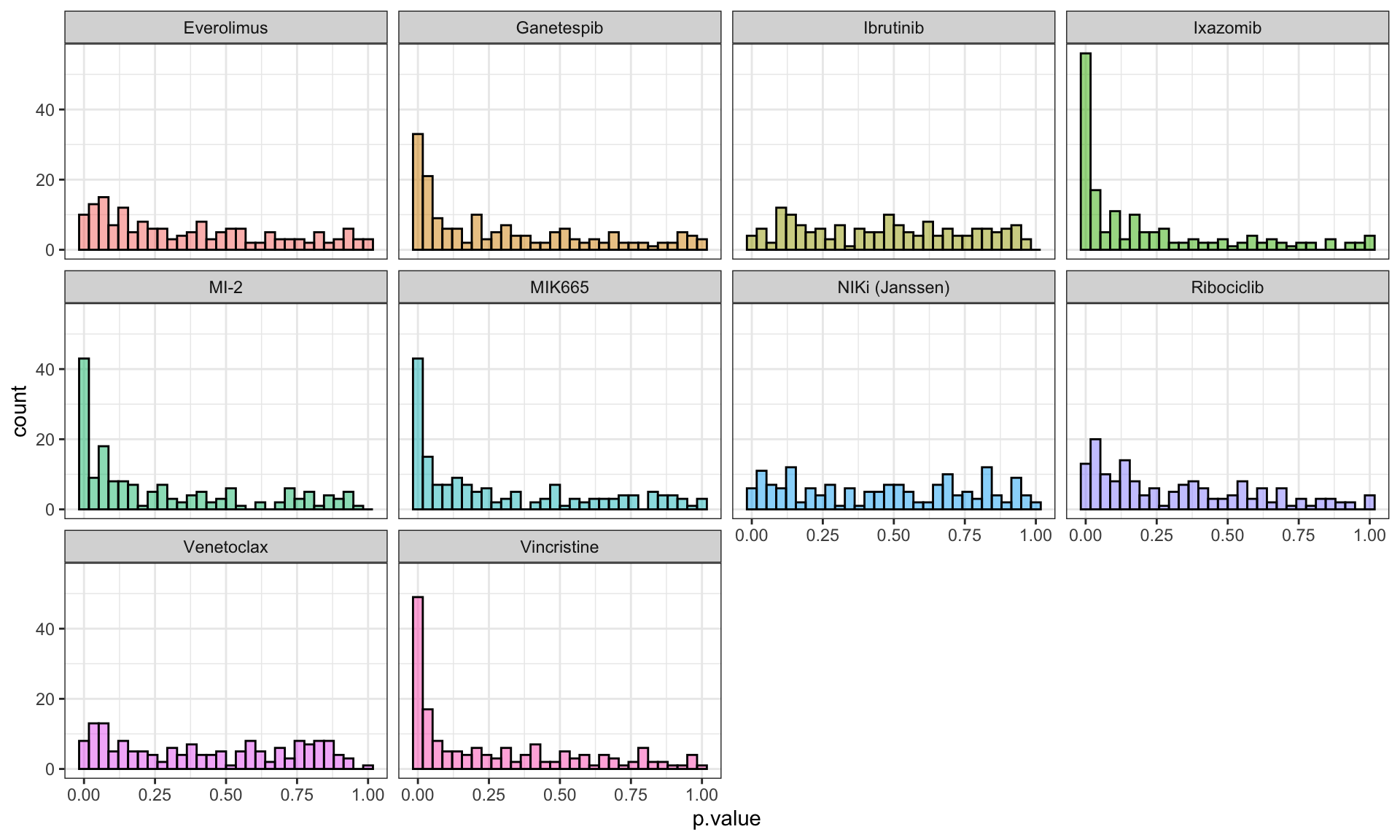
Correlation coefficient distribution
ggplot(resTab.conc, aes(x=estimate, fill = Drug_A)) +
geom_density(alpha =0.5) +
facet_wrap(~Drug_A) +
geom_vline(xintercept = 0, linetype = "dashed", color ="red") +
theme(legend.position = "none") +
xlab("Correlation coefficient")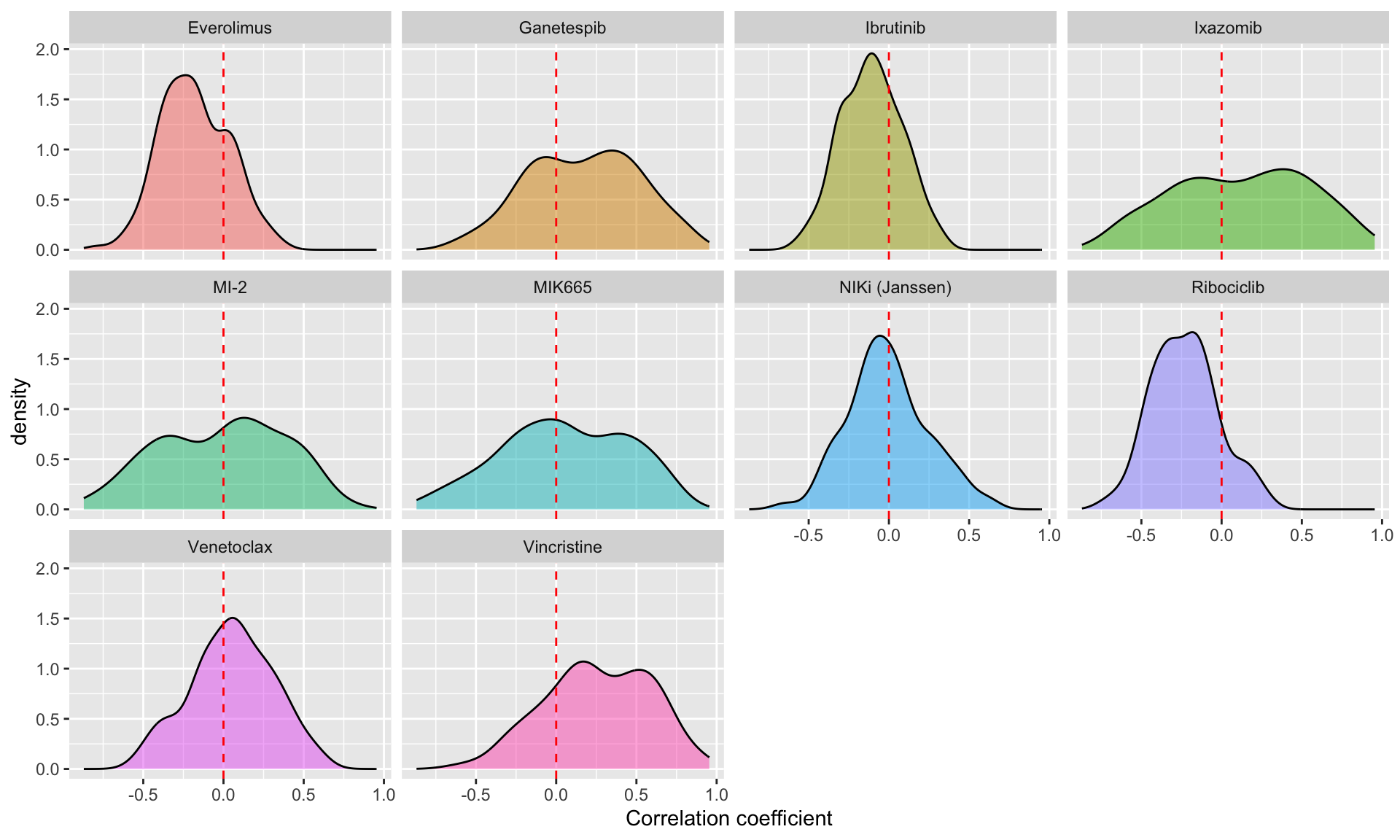
P-value heatmap
plotTab <- select(resTab.conc, Drug_A, Drug_B, Drug_A.ConcStep, estimate, p.value, p.adj) %>%
mutate(Psign = sign(estimate)*(-log10(p.value))) %>%
mutate(ifSig = ifelse(p.adj <= 0.10, "*",""))
ggplot(plotTab, aes(x=Drug_A.ConcStep, y=Drug_B, fill = Psign)) +
geom_tile() +
geom_text(aes(label = ifSig), vjust =0.5) +
scale_fill_gradient2() +
facet_wrap(~Drug_A, nrow=1)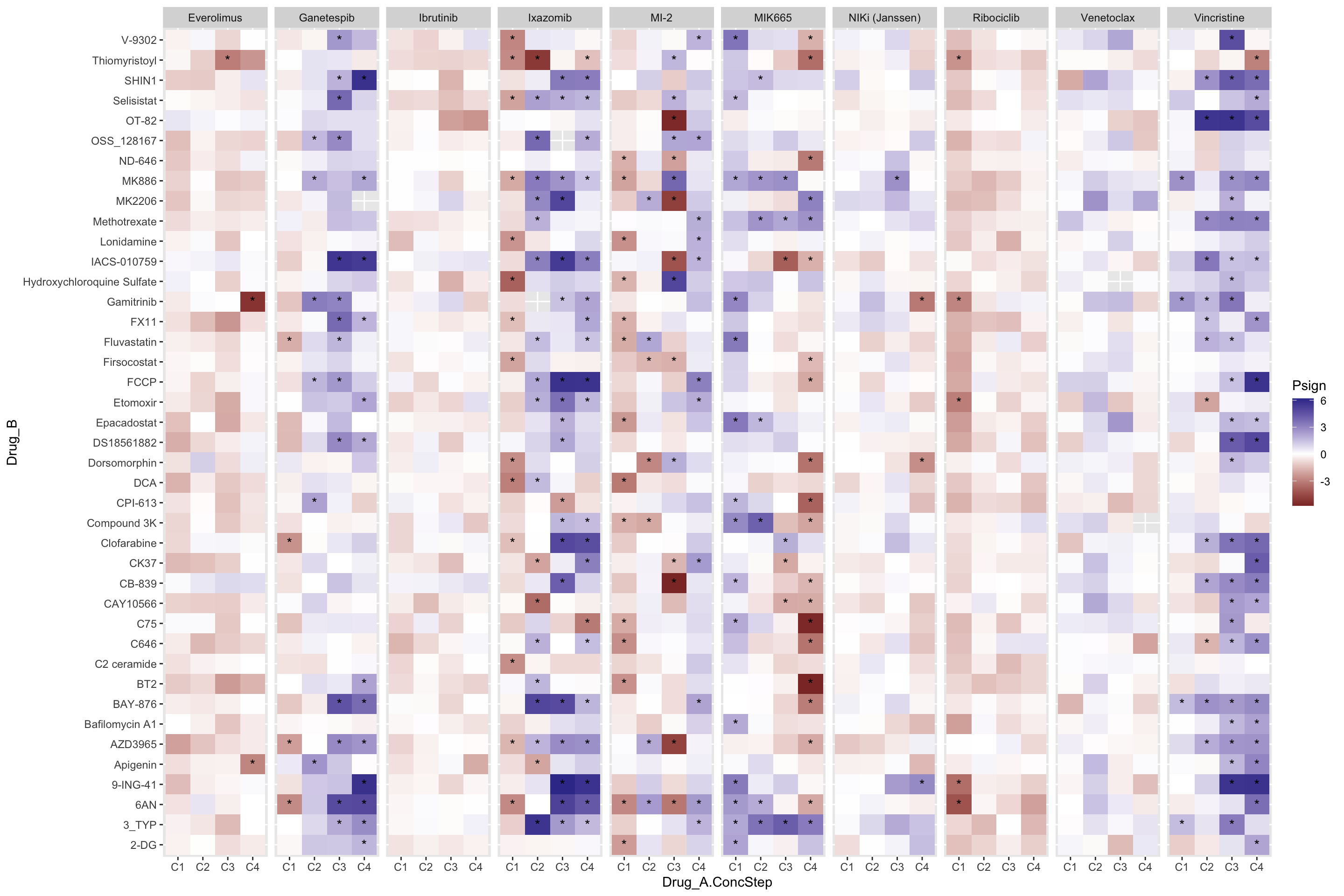
Scatter plot of significant associations
Top 20 negative associations: higher resistance correlated with lower CI (more synergistic)
plotDrug <- filter(resTab.conc, estimate <0)
pList <- lapply(seq(20), function(i) {
rec <- plotDrug[i,]
plotTab <- filter(synTab, Drug_A == rec$Drug_A, Drug_B == rec$Drug_B,
Drug_A.ConcStep == rec$Drug_A.ConcStep)
ggplot(plotTab, aes(x=viabA, y=CI)) +
geom_point() + geom_smooth(method="lm") +
ggtitle(sprintf("%s ~ %s\n(concStep: %s)", rec$Drug_A, rec$Drug_B, rec$Drug_A.ConcStep)) +
xlab(rec$Drug_A) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList, ncol=4)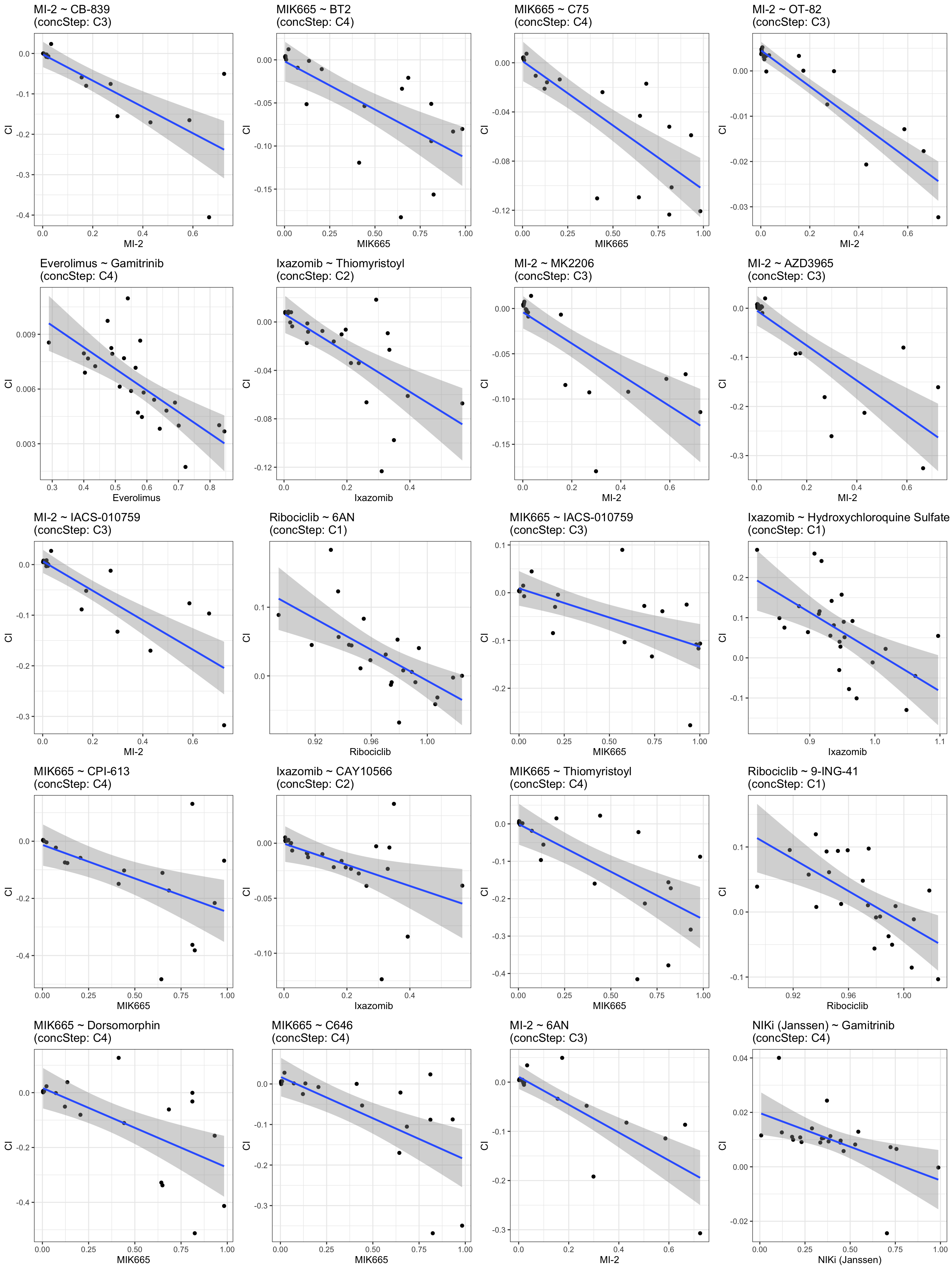
Top 9 positive associations: higher resistance correlated with higher CI (more antagonistic)
plotDrug <- filter(resTab.conc, estimate >0)
pList <- lapply(seq(20), function(i) {
rec <- plotDrug[i,]
plotTab <- filter(synTab, Drug_A == rec$Drug_A, Drug_B == rec$Drug_B,
Drug_A.ConcStep == rec$Drug_A.ConcStep)
ggplot(plotTab, aes(x=viabA, y=CI)) +
geom_point() + geom_smooth(method="lm") +
ggtitle(sprintf("%s ~ %s\n(concStep: %s)", rec$Drug_A, rec$Drug_B, rec$Drug_A.ConcStep)) +
xlab(rec$Drug_A) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList, ncol=4)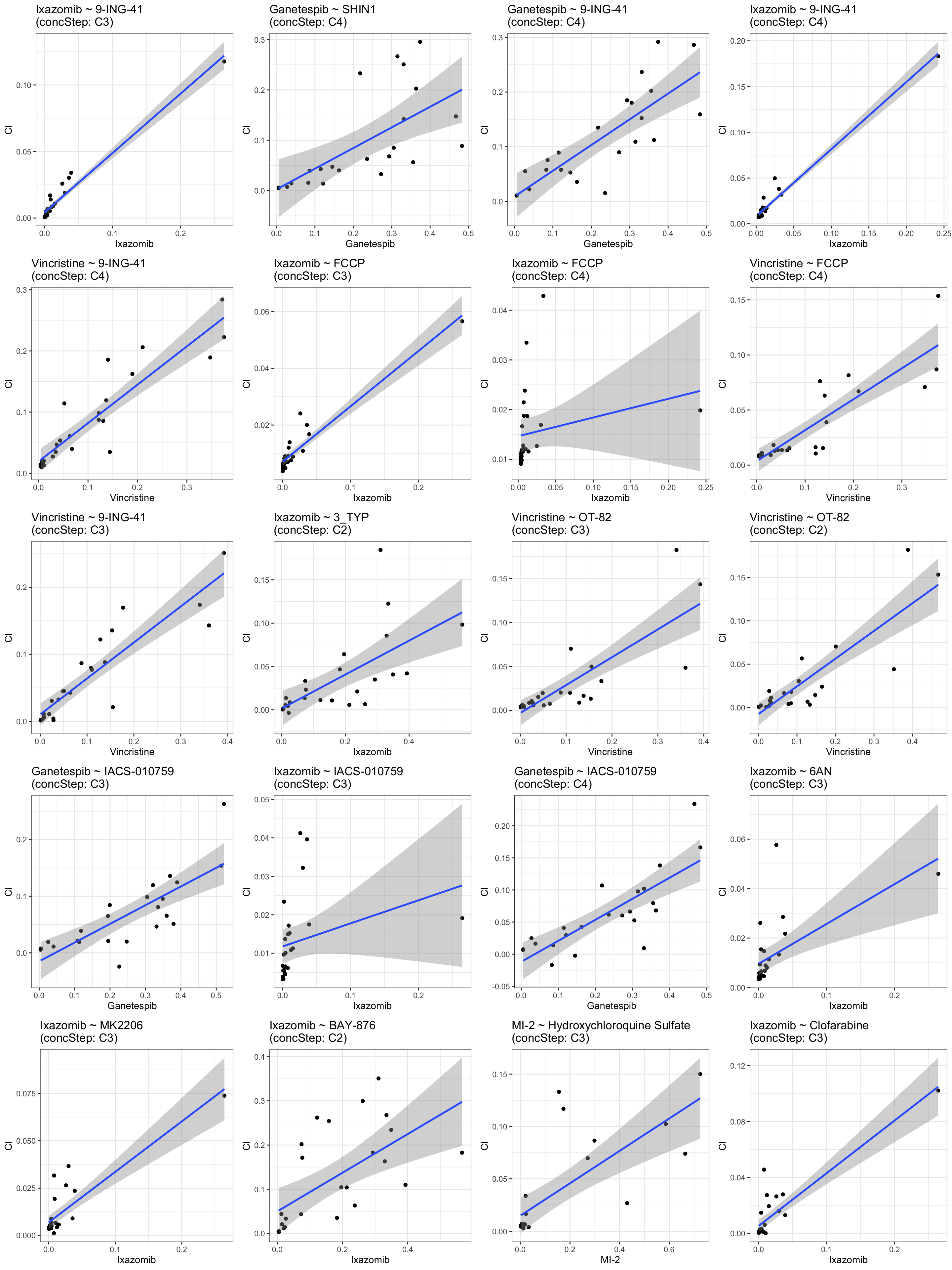
At summarised level
Use AUC for the clinical drug responses and summarised CI for combinations
Calculate AUC for clinical drugs
viabA <- drugATab %>% group_by(Name, Drug_A) %>%
summarise(auc = calcAUC(viabA, Drug_A.Conc))Correlate AUC with summarised CI
testTab <- viabA %>% left_join(ciTabSum, by = c("Drug_A","Name")) %>%
pivot_longer(c(syn, anta), names_to = "type", values_to = "score") %>%
filter(!is.na(auc), !is.na(score))
resTab.sum <- group_by(testTab, Drug_A, Drug_B, type) %>%
nest() %>%
mutate(m = map(data, ~cor.test(~auc+score,., method = "spearman"))) %>%
mutate(res = map(m, broom::tidy)) %>%
unnest(res) %>%
group_by(type, Drug_A) %>%
mutate(p.adj = p.adjust(p.value, method = "BH")) %>%
ungroup() %>% arrange(p.value)P-value histogram per clinical drug
ggplot(resTab.sum, aes(x=p.value)) +
geom_histogram(aes(fill = Drug_A), alpha=0.5, color = "black") +
facet_wrap(~Drug_A+type, scale = "free_y", ncol=4) +
theme_bw() +
theme(legend.position = "none")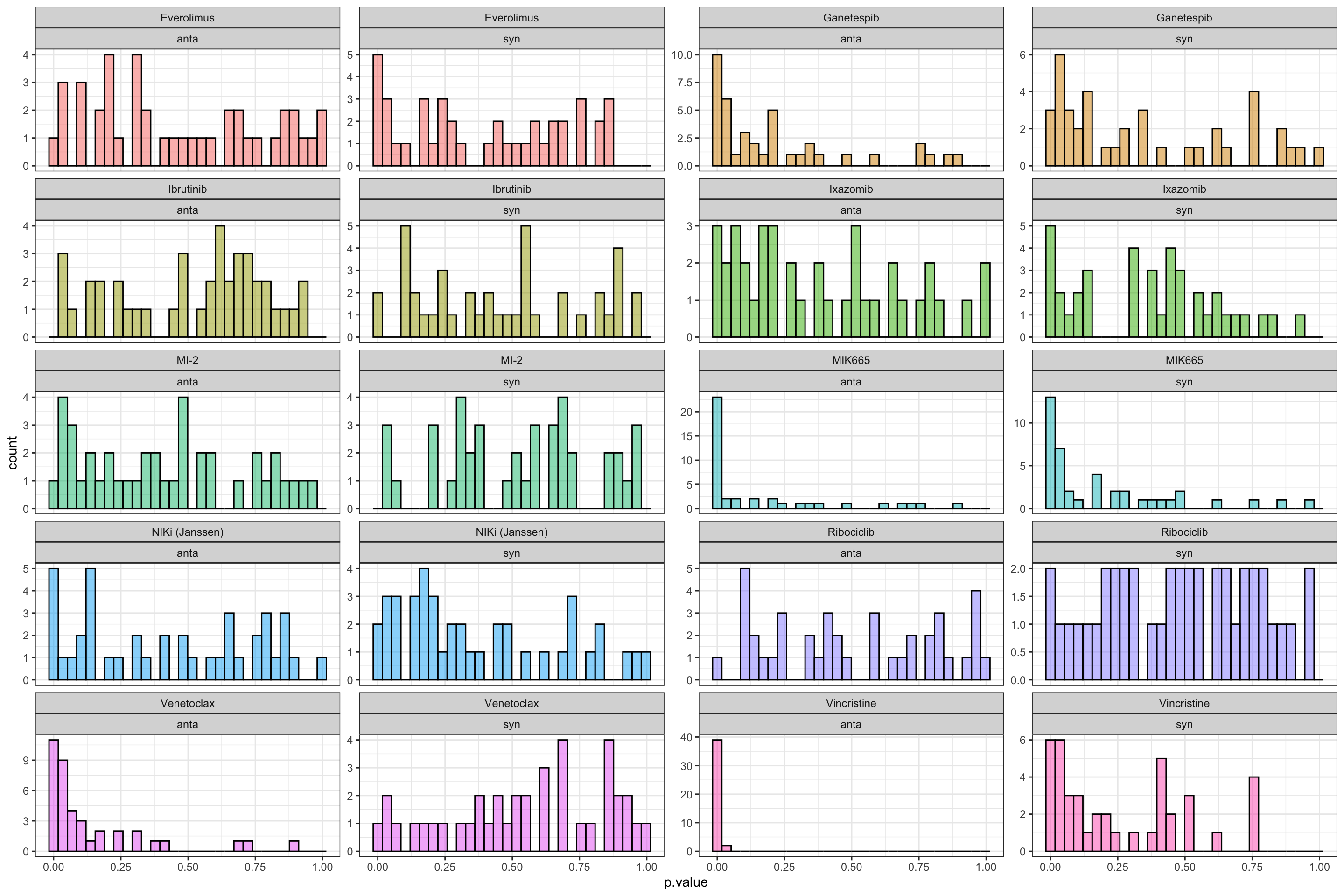
Correlation coefficient distribution
ggplot(resTab.sum, aes(x=estimate, fill = Drug_A)) +
geom_density(alpha =0.5) +
facet_wrap(~Drug_A+type, ncol=4) +
geom_vline(xintercept = 0, linetype = "dashed", color ="red") +
theme(legend.position = "none") +
xlab("Correlation coefficient")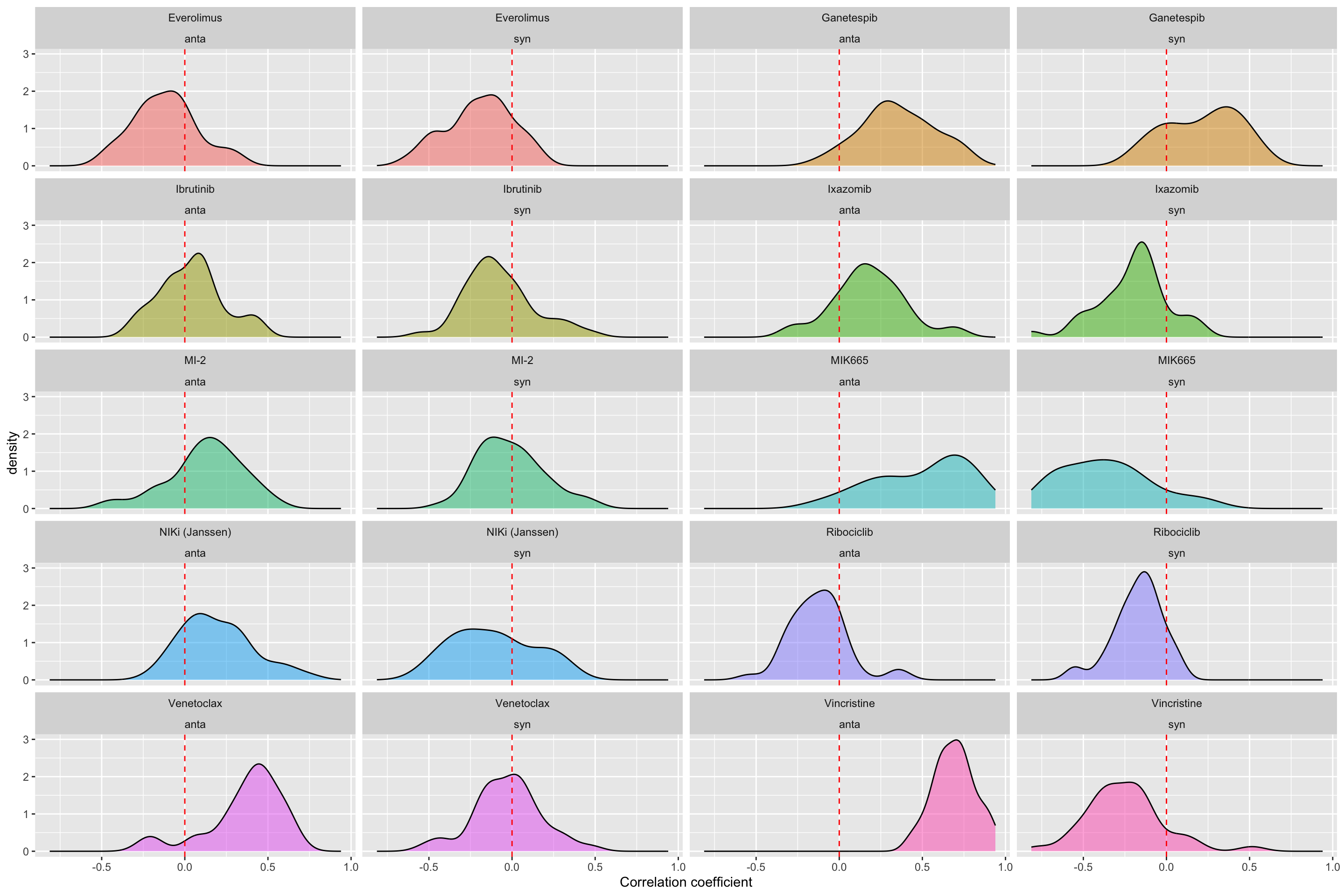
P-value heatmap
plotTab <- select(resTab.sum, Drug_A, Drug_B, estimate, p.value, p.adj,type) %>%
mutate(Psign = sign(estimate)*(-log10(p.value))) %>%
mutate(ifSig = ifelse(p.adj <= 0.10, "*",""))
ggplot(plotTab, aes(x=Drug_A, y=Drug_B, fill = Psign)) +
geom_tile() +
geom_text(aes(label = ifSig), vjust =0.5) +
scale_fill_gradient2() +
facet_wrap(~type, nrow=1) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))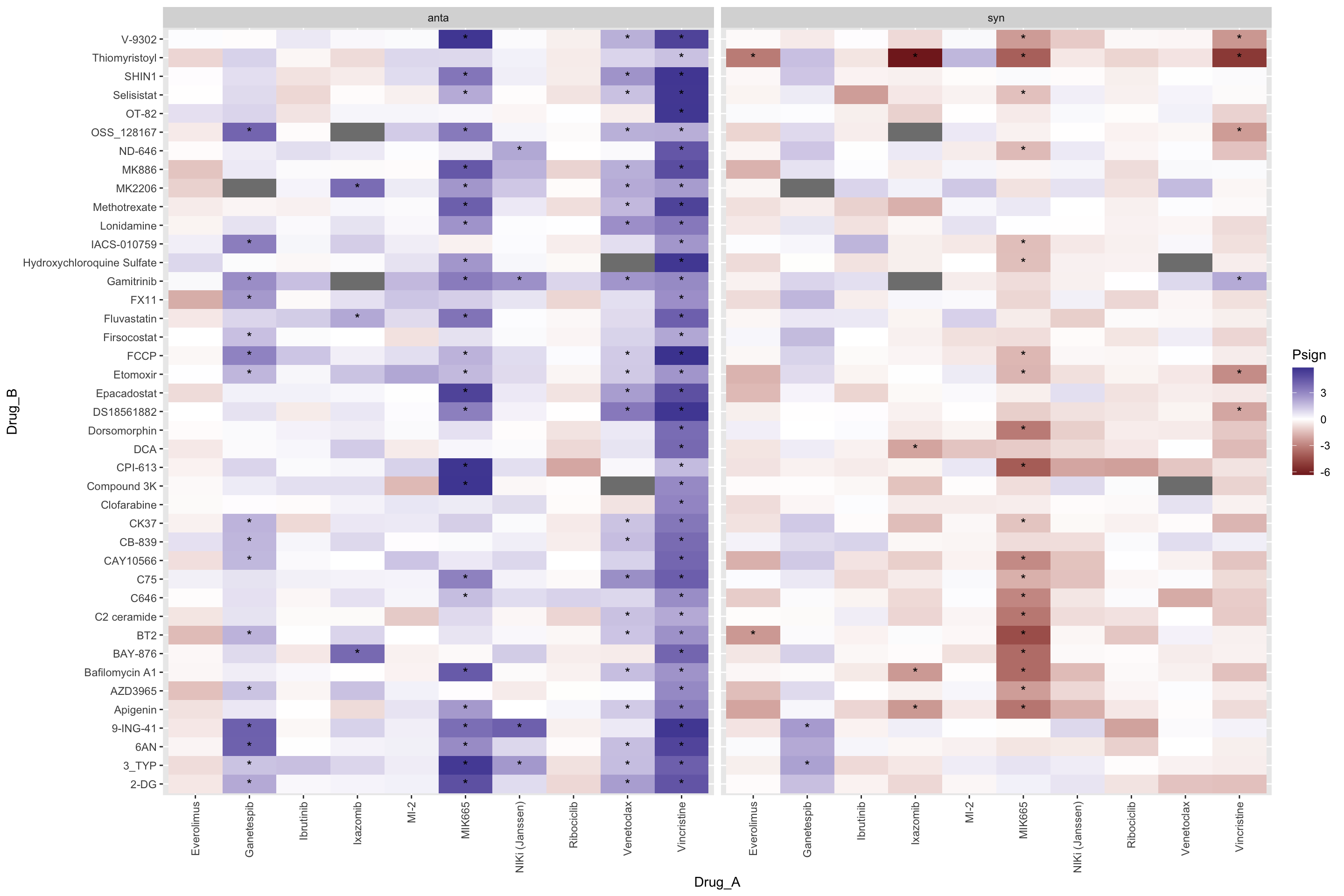
Scatter plot of significant associations
Top 20 negative associations with synergy: higher resistance correlated with higher synergy
plotDrug <- filter(resTab.sum, estimate <0, type =="syn")
pList <- lapply(seq(20), function(i) {
rec <- plotDrug[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Drug_B == rec$Drug_B,
type == rec$type)
ggplot(plotTab, aes(x=auc, y=score)) +
geom_point() + geom_smooth(method="lm") +
ggtitle(sprintf("%s ~ %s (synergy)", rec$Drug_A, rec$Drug_B)) +
xlab(rec$Drug_A) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList, ncol=4)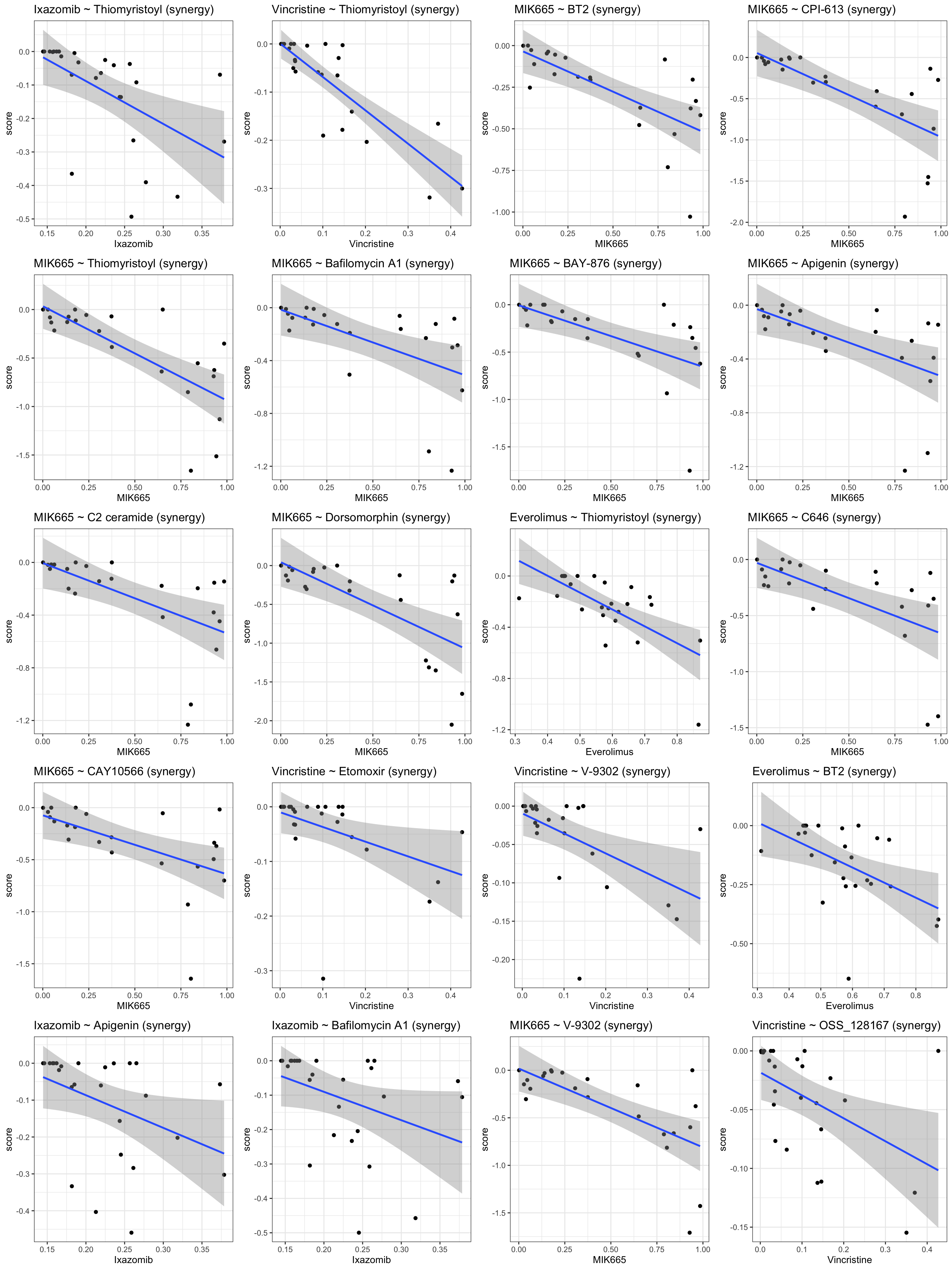
Top 20 positive associations with antagonistic: higher resistance correlated with more antagonistic effect
plotDrug <- filter(resTab.sum, estimate >0, type =="anta")
pList <- lapply(seq(20), function(i) {
rec <- plotDrug[i,]
plotTab <- filter(testTab, Drug_A == rec$Drug_A, Drug_B == rec$Drug_B,
type == rec$type)
ggplot(plotTab, aes(x=auc, y=score)) +
geom_point() + geom_smooth(method="lm") +
ggtitle(sprintf("%s ~ %s (antagonistic)", rec$Drug_A, rec$Drug_B)) +
xlab(rec$Drug_A) +
theme_bw()
})
cowplot::plot_grid(plotlist = pList, ncol=4)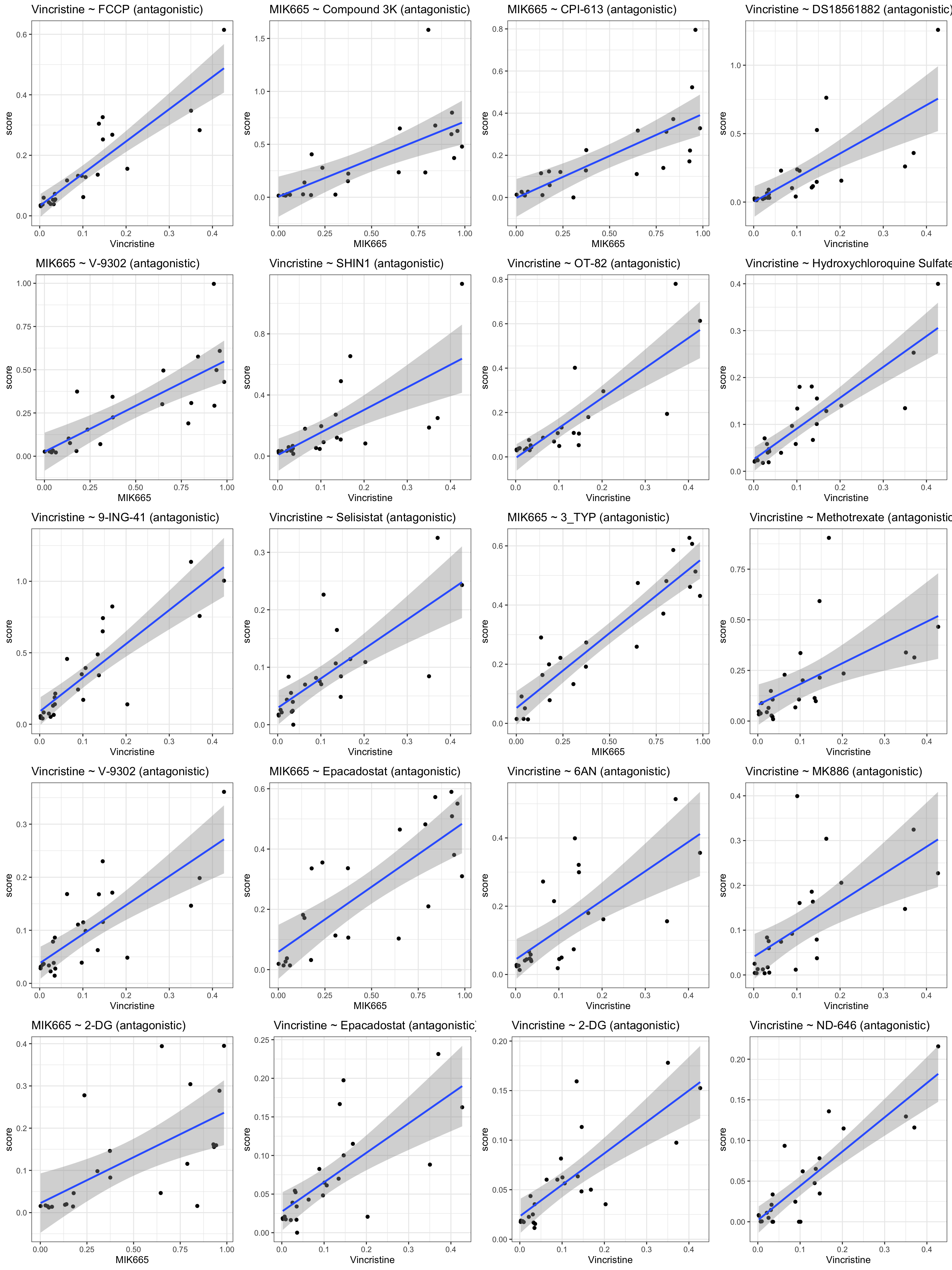
Define non-responder cell lines and test for synergy
Define non-responders to clinical drugs using the mirror method proposed by Wolfgang
Plot response distribution for each clinical drug
ggplot(viabA, aes(x=auc)) + geom_histogram() + facet_wrap(~Drug_A)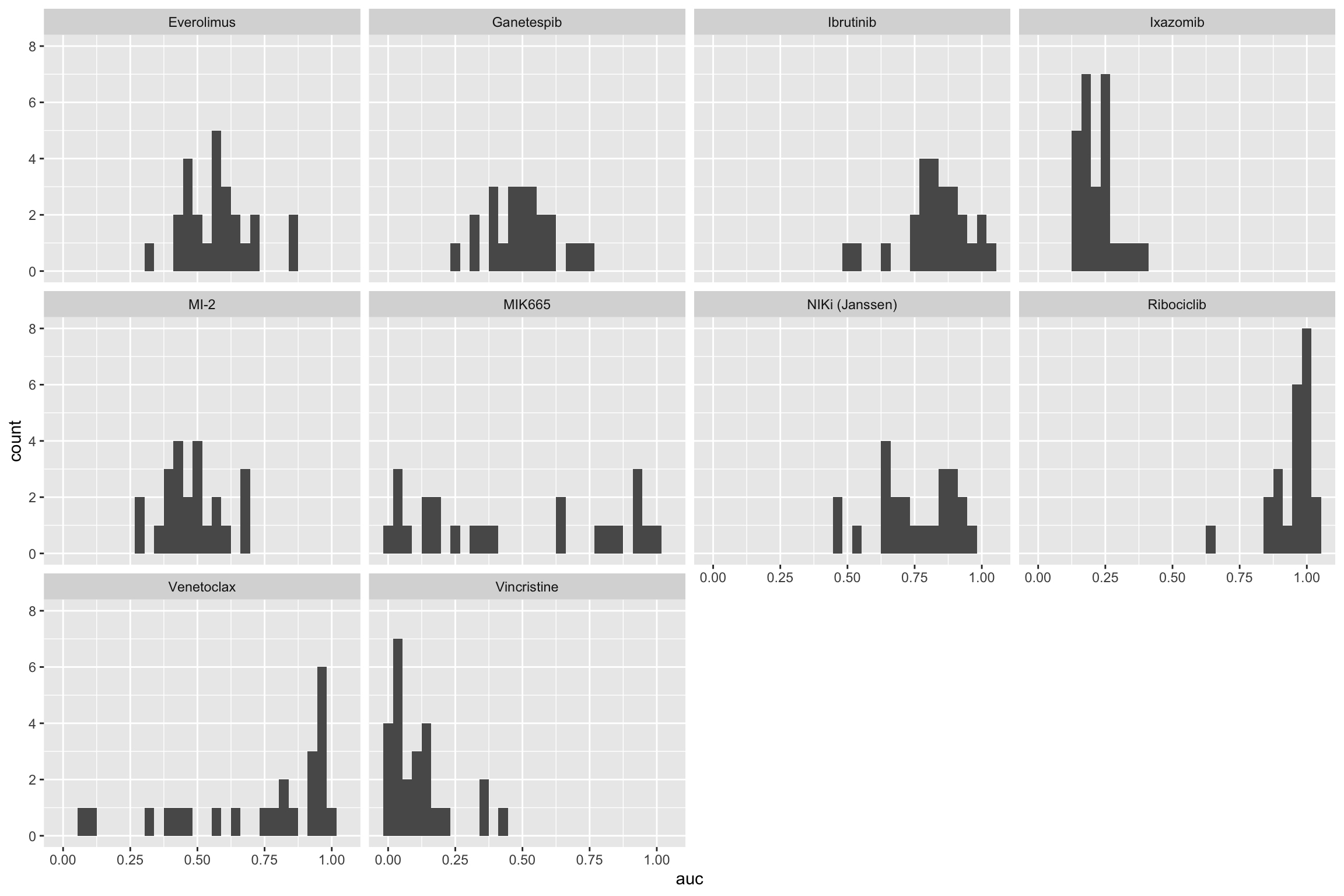 The mirro method may not work for this dataset. Perhaps it’s better to
just define the response group manually?
The mirro method may not work for this dataset. Perhaps it’s better to
just define the response group manually?
Comapre BayeSynergy score with Bliss synergy score
Load BayeSynergy score calcualted by Antonia
load("../output/fit_BayeSynergy_AA.rdata")
comTabCI <- fit$screenSummary %>%
select(`Experiment ID`, `Drug A`, `Drug B`, `Synergy Score`, `Antagonism Score`) %>%
dplyr::rename(Name = `Experiment ID`, Drug_A = `Drug A`, Drug_B = `Drug B`, syn = `Synergy Score`, anta = `Antagonism Score`) %>%
mutate(model = "BayeSynergy") %>%
bind_rows(mutate(ciTabSum, model = "Bliss")) %>%
pivot_longer(c(syn,anta), names_to = "type", values_to = "score") %>%
pivot_wider(names_from = model, values_from = score)Overall comparison
ggplot(comTabCI, aes(x=Bliss, y=BayeSynergy)) +
geom_point(aes(color = type)) +
theme_bw()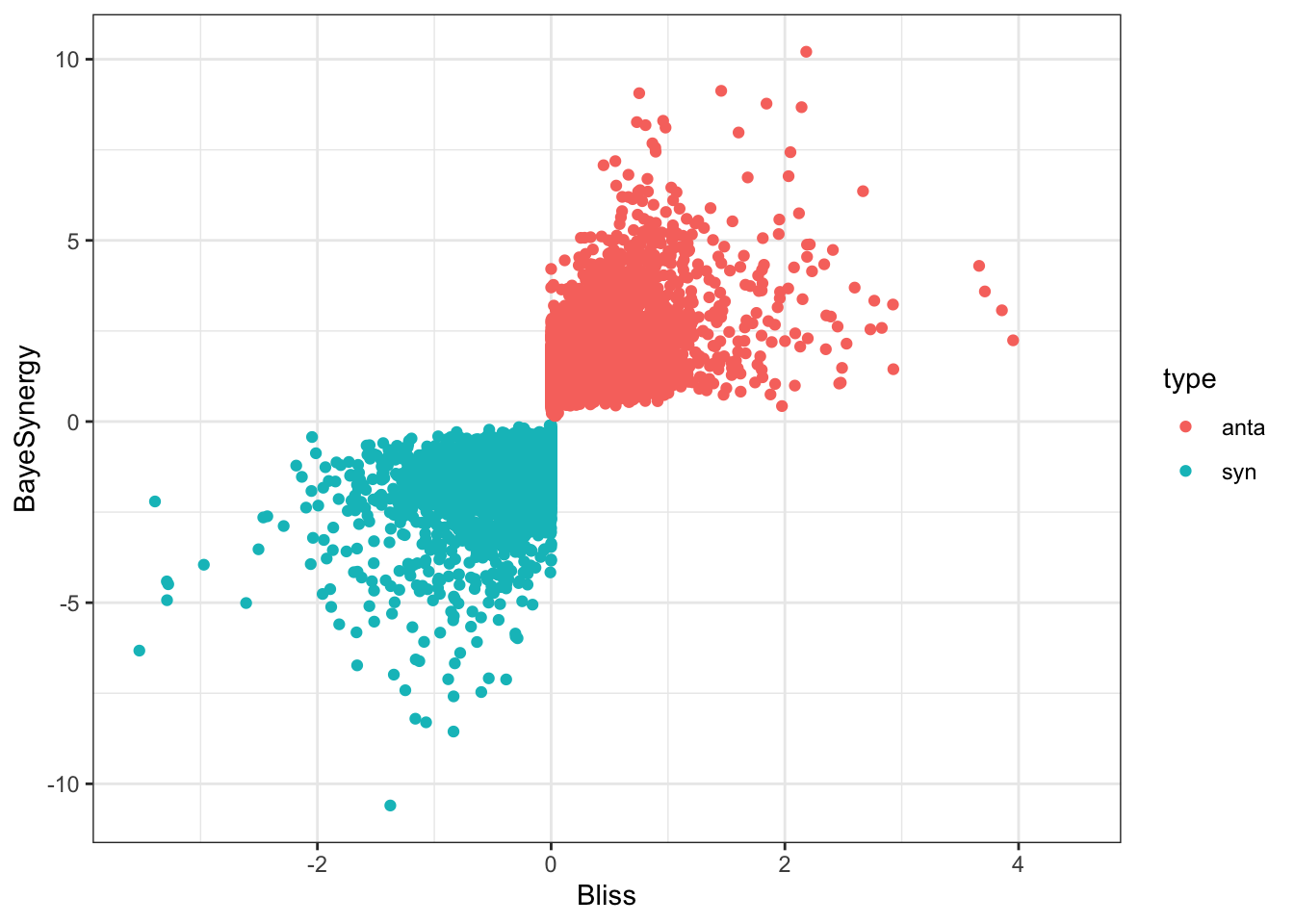 Overall the synergy and antagonistic scores from Bliss models and
Bayesynergy are comparable.
Overall the synergy and antagonistic scores from Bliss models and
Bayesynergy are comparable.
Visualize per cellline + clinical drug
pList <- lapply(unique(comTabCI$Drug_A), function(drugA) {
plotTab <- filter(comTabCI, Drug_A == drugA) %>% filter(!is.na(BayeSynergy), !is.na(Bliss))
#label the top 5 CIs by either BayeSynergy or Bliss
selectTopN <- function(tab, n=3) {
topBayes <- arrange(tab, desc(abs(BayeSynergy)))$Drug_B[seq(n)]
topBliss <- arrange(tab, desc(abs(Bliss)))$Drug_B[seq(n)]
return(tibble(drugName = unique(c(topBayes, topBliss))))
}
topDrug <- group_by(plotTab, Name, Drug_A, type) %>% nest() %>%
mutate(res = map(data, selectTopN)) %>%
unnest(res) %>%
select(-data) %>%
mutate(Drug_B = drugName)
plotTab <- left_join(plotTab, topDrug, by = c("Name","Drug_A","type","Drug_B"))
ggplot(plotTab, aes(x=Bliss, y=BayeSynergy)) +
geom_point(aes(color = type)) +
facet_wrap(~Name, ncol=5) +
ggrepel::geom_text_repel(aes(label = drugName), max.overlaps = Inf) +
theme_bw() +
geom_hline(yintercept = 0, linetype = "dashed", color = "grey50") +
geom_vline(xintercept = 0, linetype = "dashed", color = "grey50") +
ggtitle(drugA) +
theme(plot.title = element_text(hjust = 0.5,size = 20, face="bold"))
})
jyluMisc::makepdf(pList,"../docs/comapre_Bliss_BayeSynergy.pdf",ncol=1, nrow=1, width = 15, height = 15)comapre_Bliss_BayeSynergy.pdf
The top 5 synergy and antagonism from either Bliss or Bayesynergy are
labeled.
Multi-variate regression to identfy associations between genomics and synergy
Bliss synergy
Prepare genomic table
mutMat <- mutTab %>% pivot_wider(names_from = Gene, values_from = status) Selected drug pairs for visualization
selectedPair <- tibble(Drug_A = c("MI-2","MI-2","MIK665","MIK665","Ixazomib","Ixazomib"),
Drug_B = c("AZD3965","IACS-010759","AZD3965","IACS-010759","BT2","C2 ceramide"))Multi-variate linear regression for each combination
drugPair <- ungroup(ciTabSum) %>% distinct(Drug_A, Drug_B) #test for all drug pairs
allRes <- BiocParallel::bplapply(seq(nrow(drugPair)), function(i) {
rec <- drugPair[i,]
synVal <- filter(ciTabSum, Drug_A == rec$Drug_A, Drug_B==rec$Drug_B) %>%
ungroup() %>%
select(Name, syn)
testTab <- left_join(mutMat, synVal, by = "Name") %>%
column_to_rownames("Name")
m <- summary(lm(syn ~ ., testTab))
R2 <- m$r.squared
coefTab <- m$coefficients[-1,c(1,4)] %>% data.frame() %>%
rownames_to_column("Gene") %>%
as_tibble() %>%
dplyr::rename(p = "Pr...t..")
#colnames(coefTab) <- c("coef","p")
tibble(Drug_A = rec$Drug_A, Drug_B = rec$Drug_B, R2 = R2, coefTab = list(coefTab)) %>%
mutate(R2 = replace_na(R2,0))
}) %>%
bind_rows() %>% arrange(desc(R2)) %>% mutate(combination = paste0(Drug_A,"_", Drug_B))
seleRes <- allRes %>% filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B))Variance explained (R2) for each selected synergy by genomics
plotTab <- seleRes %>%
mutate(combination = factor(combination, levels = combination))
ggplot(plotTab, aes(x=combination, y=R2)) +
geom_bar(aes(fill = combination), stat = "identity") +
coord_flip() +
theme(legend.position = "none")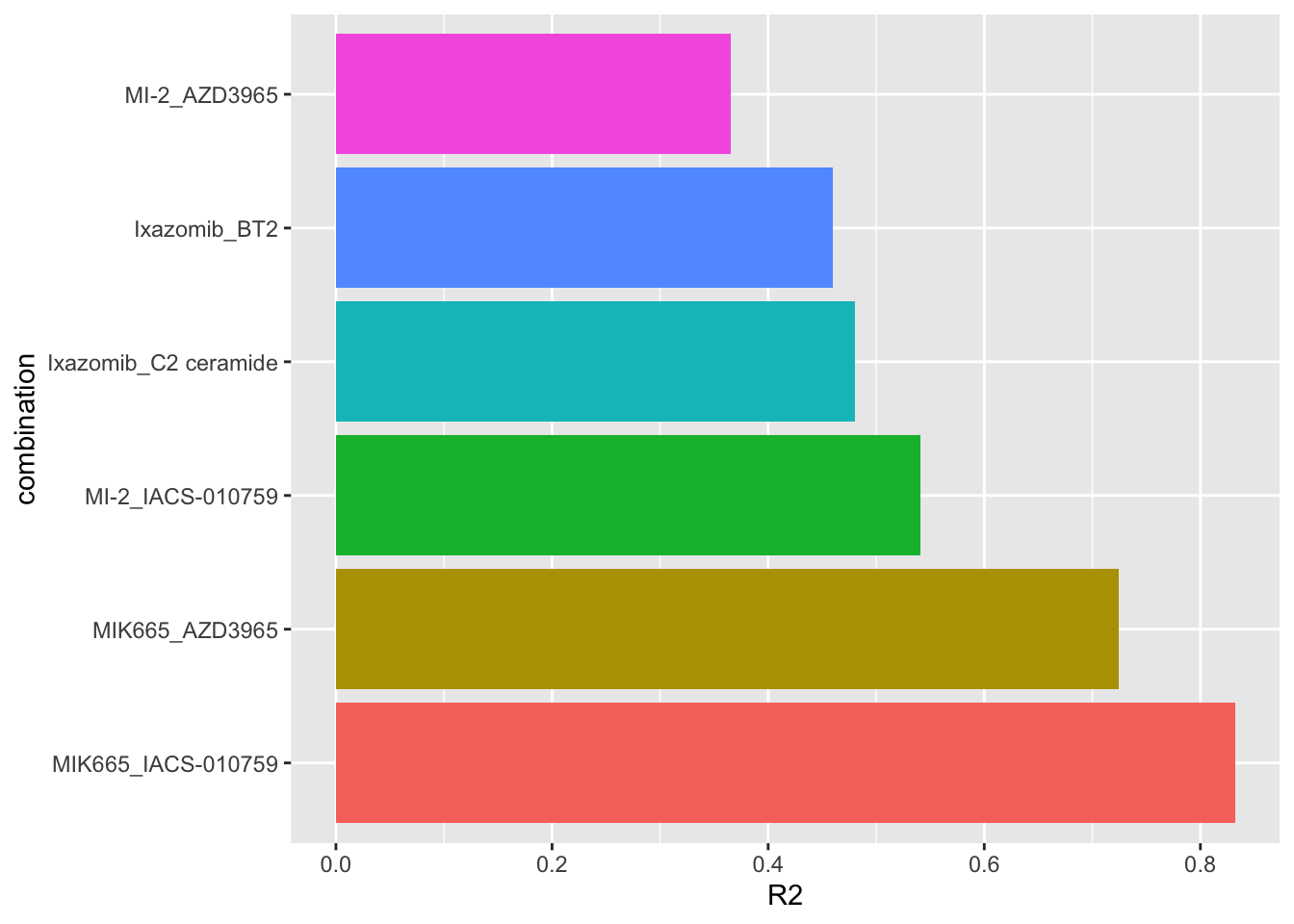
Genes that can explain synergy in multi-variate models (for selected drug pairs)
plotTab <- unnest(seleRes, coefTab) %>%
mutate(ifSig = ifelse(p <= 0.05, ifelse(p<=0.01,"**","*"),"")) %>%
mutate(combination = sprintf("%s_%s (R2=%1.1f)",Drug_A, Drug_B, R2))
ggplot(plotTab, aes(x=Gene, y=combination, fill = Estimate)) +
geom_tile() +
geom_text(aes(label = ifSig)) +
scale_fill_gradient2(low = "blue", high = "red", mid="white", midpoint = 0, name ="coefficient")+
ggtitle("Selected drug pairs") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5),
plot.title = element_text(face = "bold", hjust = 0.5)) 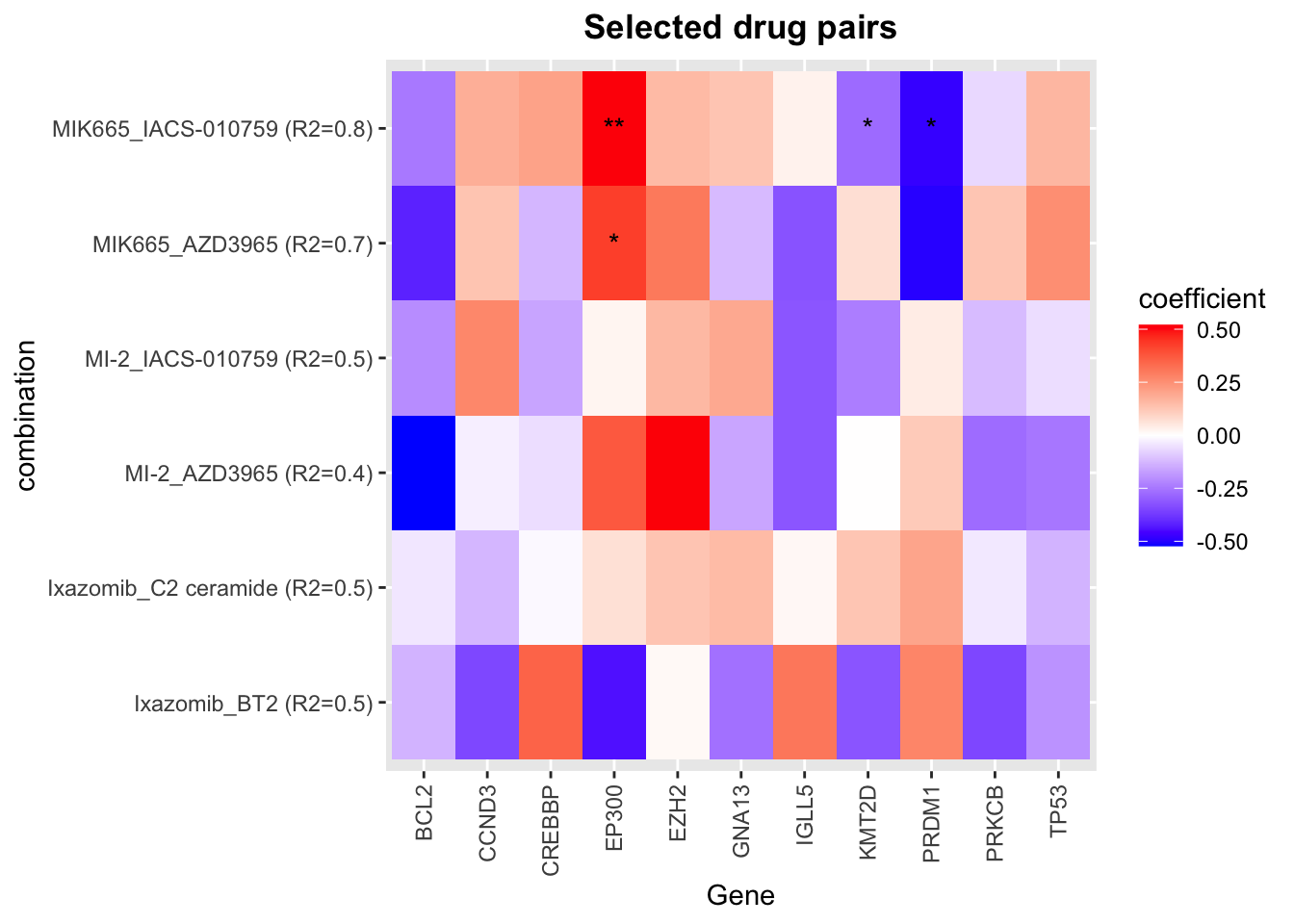
Color of the heatmap indicates the coefficients of the genomic features in the multi-variate regression model to explain the CI scores. Since higher CI score is associated with higher synergy, negative coefficient (blue tiles) indicates the mutations associate with higher synergy and positive coefficient (red tiles) indicates mutations associate with lower synergy.
R2 values indicate how much variance in the synergistic scores can be explained by the genomics
* indicates the significance of the genomic features in the multi-variate models. One star is p < 0.05 and two starts indicate p<0.01.
Genes that can explain synergy in multi-variate models (for all drug pairs)
pList <- lapply(unique(allRes$Drug_A), function(name) {
plotTab <- filter(allRes, Drug_A == name) %>%
unnest(coefTab) %>%
mutate(ifSig = ifelse(p <= 0.05, ifelse(p<=0.01,"**","*"),"")) %>%
mutate(combination = sprintf("%s_%s (R2=%1.1f)",Drug_A, Drug_B, R2)) %>%
arrange(R2) %>% mutate(combination = factor(combination, levels = unique(combination)))
ggplot(plotTab, aes(x=Gene, y=combination, fill = Estimate)) +
geom_tile() +
geom_text(aes(label = ifSig)) +
scale_fill_gradient2(low = "blue", high = "red", mid="white", midpoint = 0, name ="coefficient")+
ggtitle(name) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5),
plot.title = element_text(face = "bold", hjust = 0.5))
})
jyluMisc::makepdf(pList, "../docs/multiVariate_GeneAssociation_Bliss.pdf",
ncol = 1, nrow = 1,
height = 10, width = 10)Bayes synergy
load("../output/fit_BayeSynergy_AA.rdata")
bayesTabSum <- fit$screenSummary %>%
select(`Experiment ID`, `Drug A`, `Drug B`, `Synergy Score`, `Antagonism Score`) %>%
dplyr::rename(Name = `Experiment ID`, Drug_A = `Drug A`, Drug_B = `Drug B`, syn = `Synergy Score`, anta = `Antagonism Score`)Multi-variate linear regression for each combination
drugPair <- ungroup(bayesTabSum) %>% distinct(Drug_A, Drug_B) #test for all drug pairs
allRes <- BiocParallel::bplapply(seq(nrow(drugPair)), function(i) {
rec <- drugPair[i,]
synVal <- filter(bayesTabSum, Drug_A == rec$Drug_A, Drug_B==rec$Drug_B) %>%
ungroup() %>%
select(Name, syn)
testTab <- left_join(mutMat, synVal, by = "Name") %>%
column_to_rownames("Name")
m <- summary(lm(syn ~ ., testTab))
R2 <- m$r.squared
coefTab <- m$coefficients[-1,c(1,4)] %>% data.frame() %>%
rownames_to_column("Gene") %>%
as_tibble() %>%
dplyr::rename(p = "Pr...t..")
#colnames(coefTab) <- c("coef","p")
tibble(Drug_A = rec$Drug_A, Drug_B = rec$Drug_B, R2 = R2, coefTab = list(coefTab)) %>%
mutate(R2 = replace_na(R2,0))
}) %>%
bind_rows() %>% arrange(desc(R2)) %>% mutate(combination = paste0(Drug_A,"_", Drug_B))
seleRes <- allRes %>% filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B))Variance explained (R2) for each selected synergy by genomics
plotTab <- seleRes %>%
mutate(combination = factor(combination, levels = combination))
ggplot(plotTab, aes(x=combination, y=R2)) +
geom_bar(aes(fill = combination), stat = "identity") +
coord_flip() +
theme(legend.position = "none")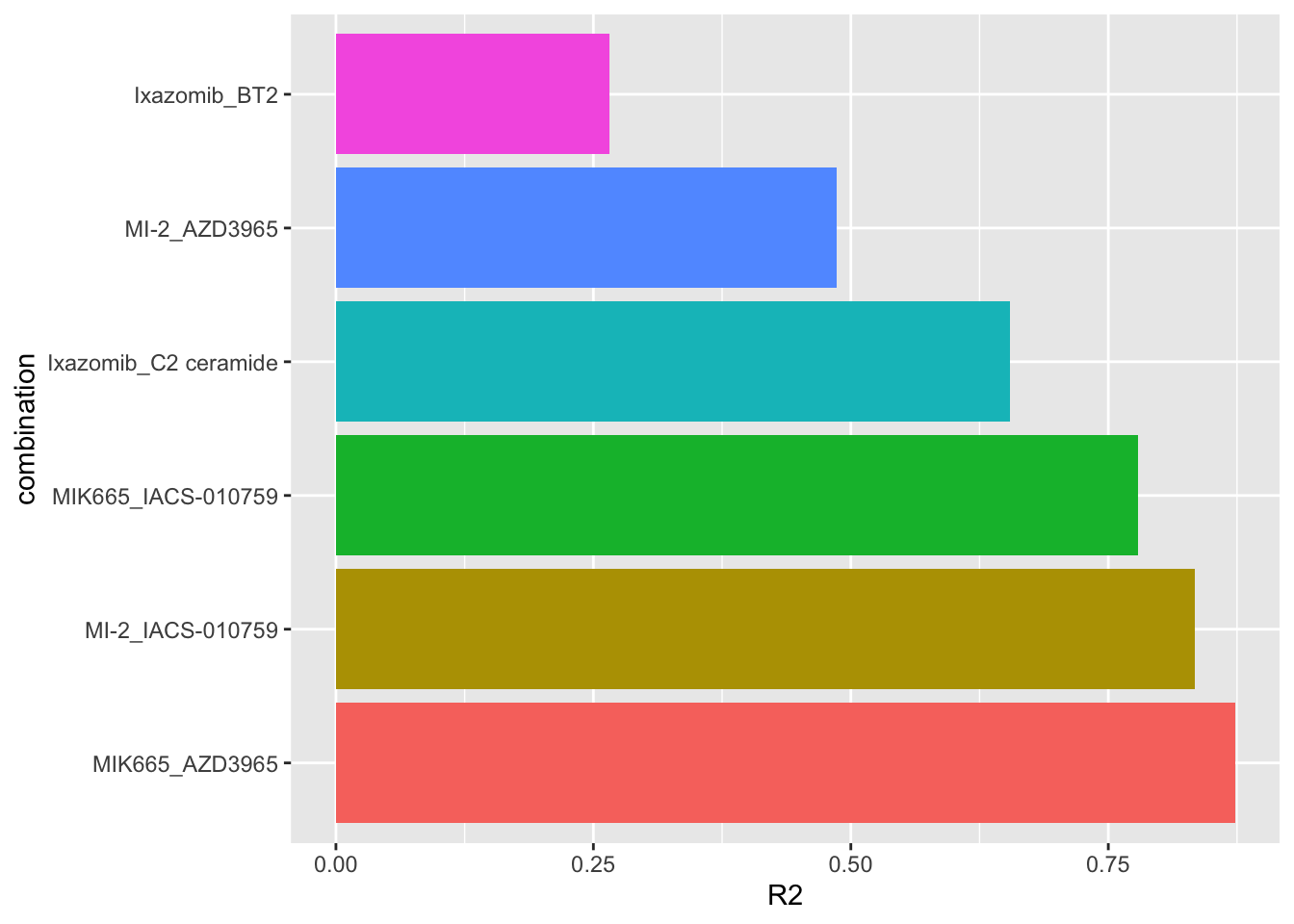
Genes that can explain synergy in multi-variate models (for selected drug pairs)
plotTab <- unnest(seleRes, coefTab) %>%
mutate(ifSig = ifelse(p <= 0.05, ifelse(p<=0.01,"**","*"),"")) %>%
mutate(combination = sprintf("%s_%s (R2=%1.1f)",Drug_A, Drug_B, R2))
ggplot(plotTab, aes(x=Gene, y=combination, fill = Estimate)) +
geom_tile() +
geom_text(aes(label = ifSig)) +
scale_fill_gradient2(low = "blue", high = "red", mid="white", midpoint = 0, name ="coefficient")+
ggtitle("Selected drug pairs") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5),
plot.title = element_text(face = "bold", hjust = 0.5)) 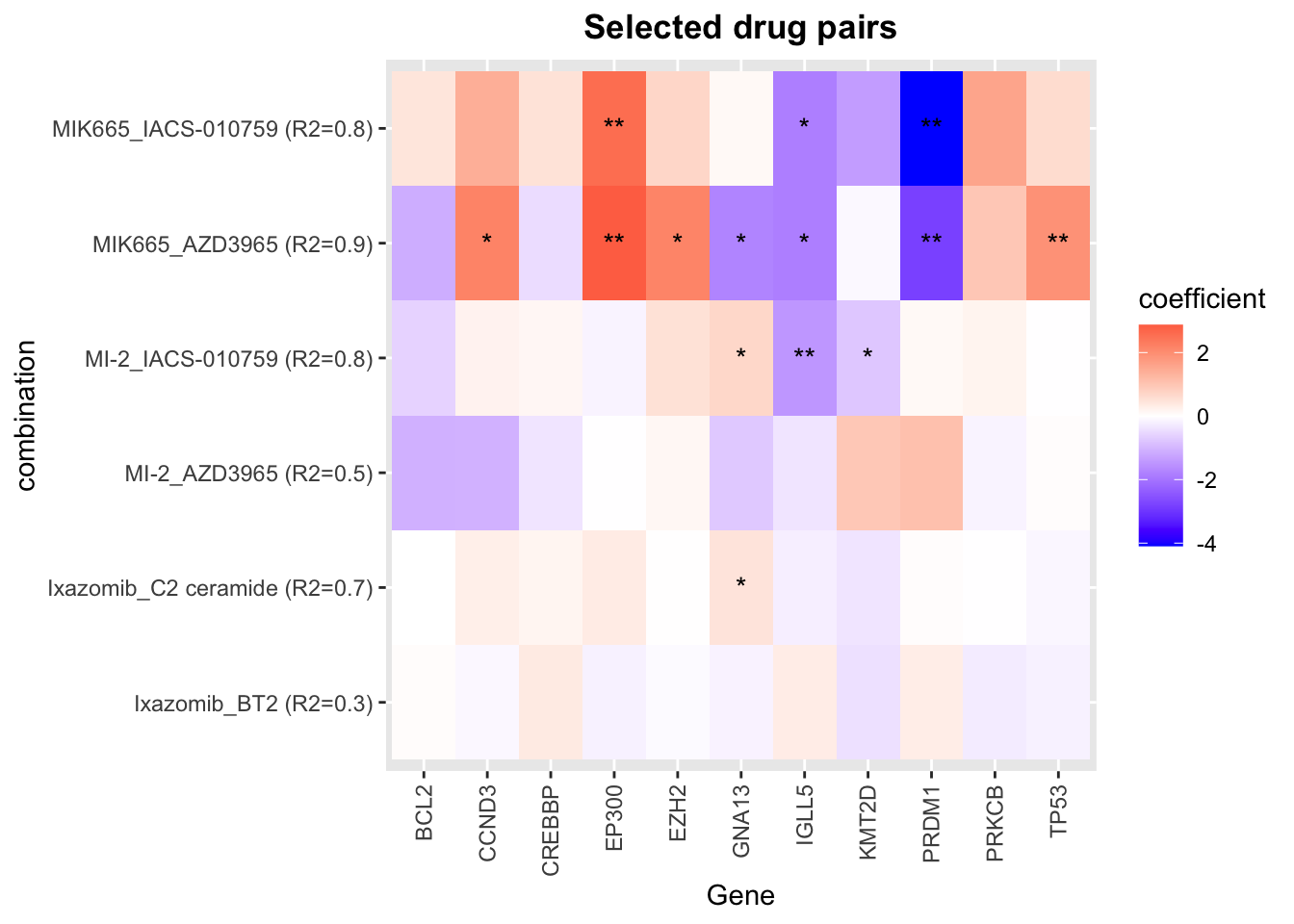
Color of the heatmap indicates the coefficients of the genomic features in the multi-variate regression model to explain the CI scores. Since higher CI score is associated with higher synergy, negative coefficient (blue tiles) indicates the mutations associate with higher synergy and positive coefficient (red tiles) indicates mutations associate with lower synergy.
R2 values indicate how much variance in the synergistic scores can be explained by the genomics
* indicates the significance of the genomic features in the multi-variate models. One star is p < 0.05 and two starts indicate p<0.01.
Genes that can explain synergy in multi-variate models (for all drug pairs)
pList <- lapply(unique(allRes$Drug_A), function(name) {
plotTab <- filter(allRes, Drug_A == name) %>%
unnest(coefTab) %>%
mutate(ifSig = ifelse(p <= 0.05, ifelse(p<=0.01,"**","*"),"")) %>%
mutate(combination = sprintf("%s_%s (R2=%1.1f)",Drug_A, Drug_B, R2)) %>%
arrange(R2) %>% mutate(combination = factor(combination, levels = unique(combination)))
ggplot(plotTab, aes(x=Gene, y=combination, fill = Estimate)) +
geom_tile() +
geom_text(aes(label = ifSig)) +
scale_fill_gradient2(low = "blue", high = "red", mid="white", midpoint = 0, name ="coefficient")+
ggtitle(name) +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5),
plot.title = element_text(face = "bold", hjust = 0.5))
})
jyluMisc::makepdf(pList, "../docs/multiVariate_GeneAssociation_Bayesynergy.pdf",
ncol = 1, nrow = 1,
height = 10, width = 10)Explain synergy score by transcriptomics
Processing transcriptomic data
Number of genes and cell lines
load("../data/DepMap_GEXwide.RData")
exprMat <- t(DepMap_GEXwide)
exprMat <- exprMat[,colnames(exprMat) %in% unique(ciTabSum$Name)]
# Remove low count genes
exprMat <- exprMat[matrixStats::rowMedians(exprMat) >0,]
vstObj <- vsn::vsnMatrix(exprMat)
exprMat <- vsn::predict(vstObj, exprMat)
dim(exprMat)[1] 15631 12Using Bliss synergy
Prepare list of selected synergy scores
synList <- ciTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination)
Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 0
2 Ixazomib_C2 ceramide 0
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 0
5 MIK665_AZD3965 179
6 MIK665_IACS-010759 0Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 87
2 Ixazomib_C2 ceramide 79
3 MI-2_AZD3965 86
4 MI-2_IACS-010759 315
5 MIK665_AZD3965 494
6 MIK665_IACS-010759 243List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Note that since more negative CI scores indicate higher synergy, genes with negative logFCs are the genes whose expressions are positively correlated with synergy
Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)Cancer Hallmark (10% FDR)
enrichres$Hallmark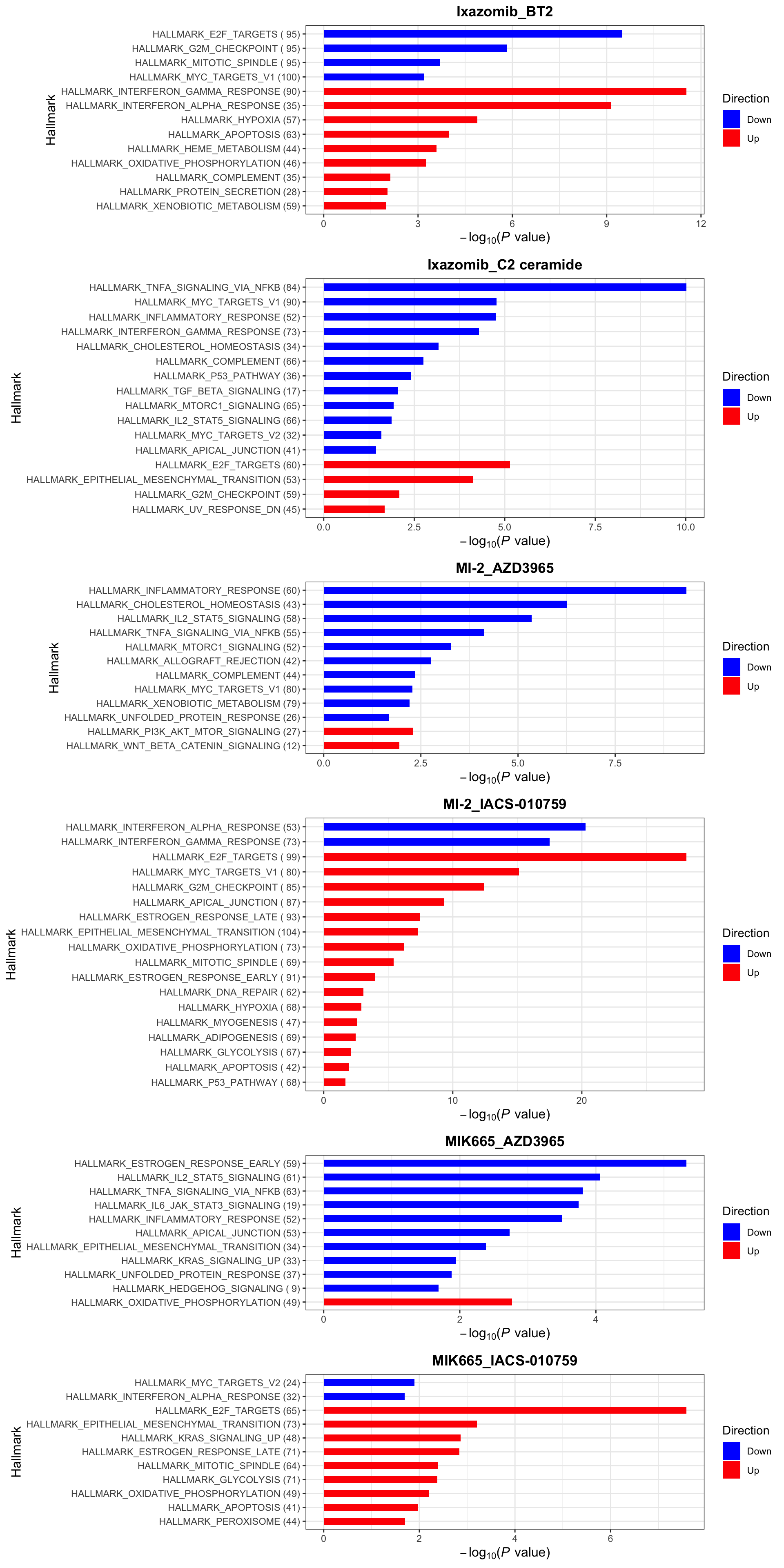 Note that since more negative CI scores indicate higher synergy,
pathways labelled as Down are the pathways associated with higher
synergy
Note that since more negative CI scores indicate higher synergy,
pathways labelled as Down are the pathways associated with higher
synergy
If a combination is not shown here, it means there’s no
significantly enriched pathways with indicated threshold
KEGG (10% FDR)
enrichres$KEGG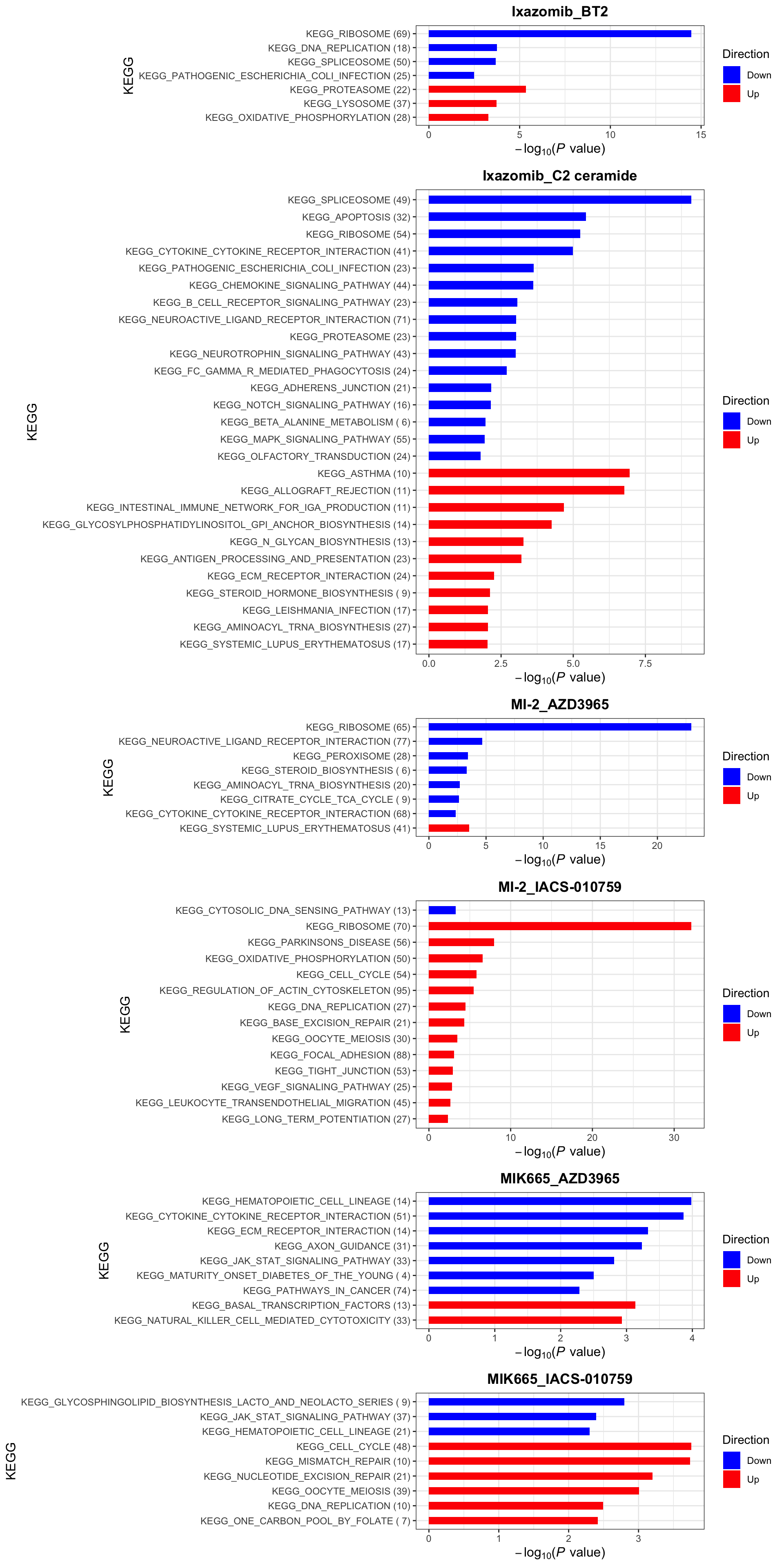 Note that since more negative CI scores indicate higher synergy,
pathways labelled as Down are the pathways associated with higher
synergy
Note that since more negative CI scores indicate higher synergy,
pathways labelled as Down are the pathways associated with higher
synergy
Using Bayes synergy
Prepare list of selected synergy scores
synList <- bayesTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination)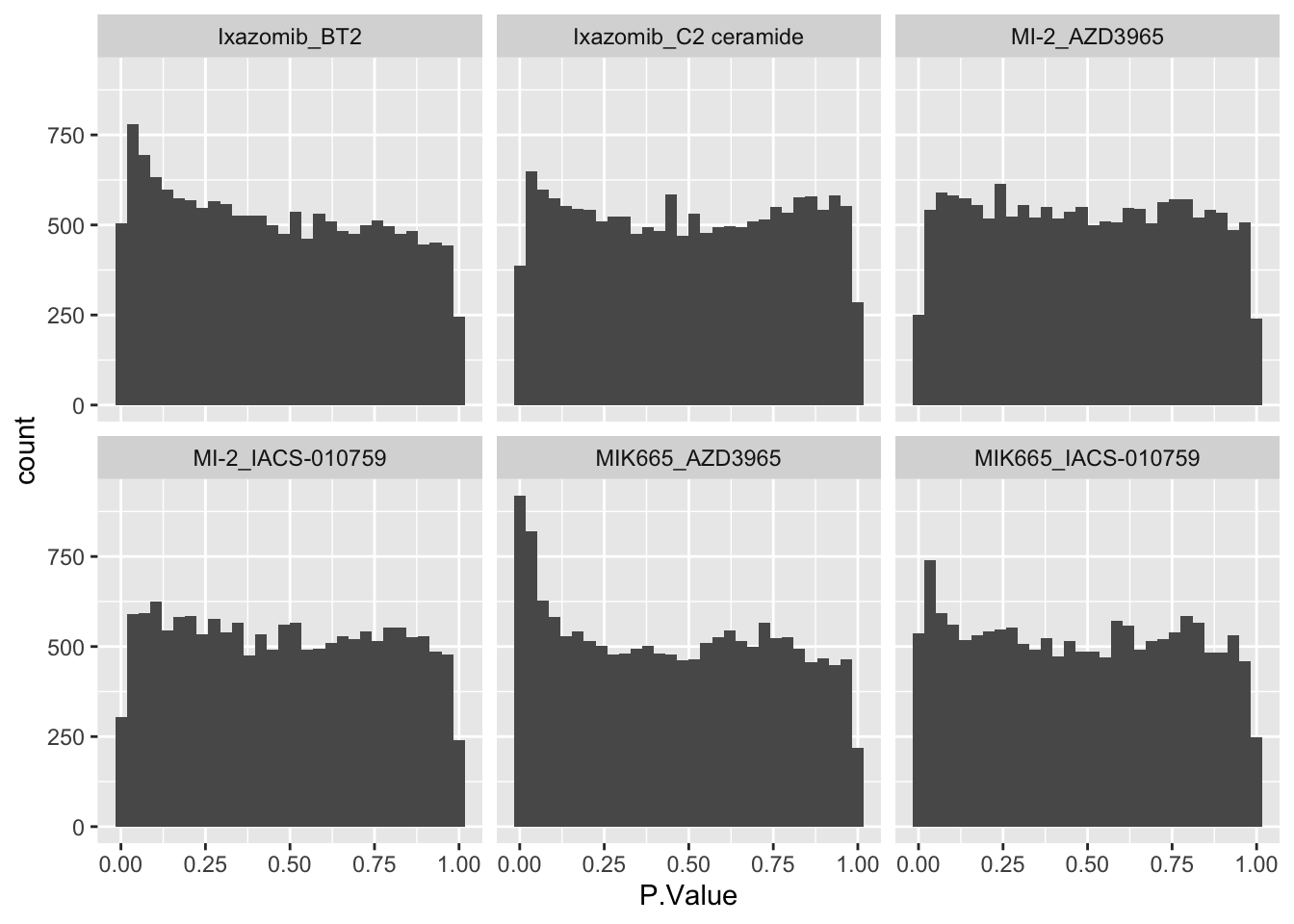 ### Number of significant associations
### Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 0
2 Ixazomib_C2 ceramide 2
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 0
5 MIK665_AZD3965 316
6 MIK665_IACS-010759 5Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 338
2 Ixazomib_C2 ceramide 218
3 MI-2_AZD3965 130
4 MI-2_IACS-010759 168
5 MIK665_AZD3965 714
6 MIK665_IACS-010759 371List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)Cancer Hallmark (10% FDR)
enrichres$Hallmark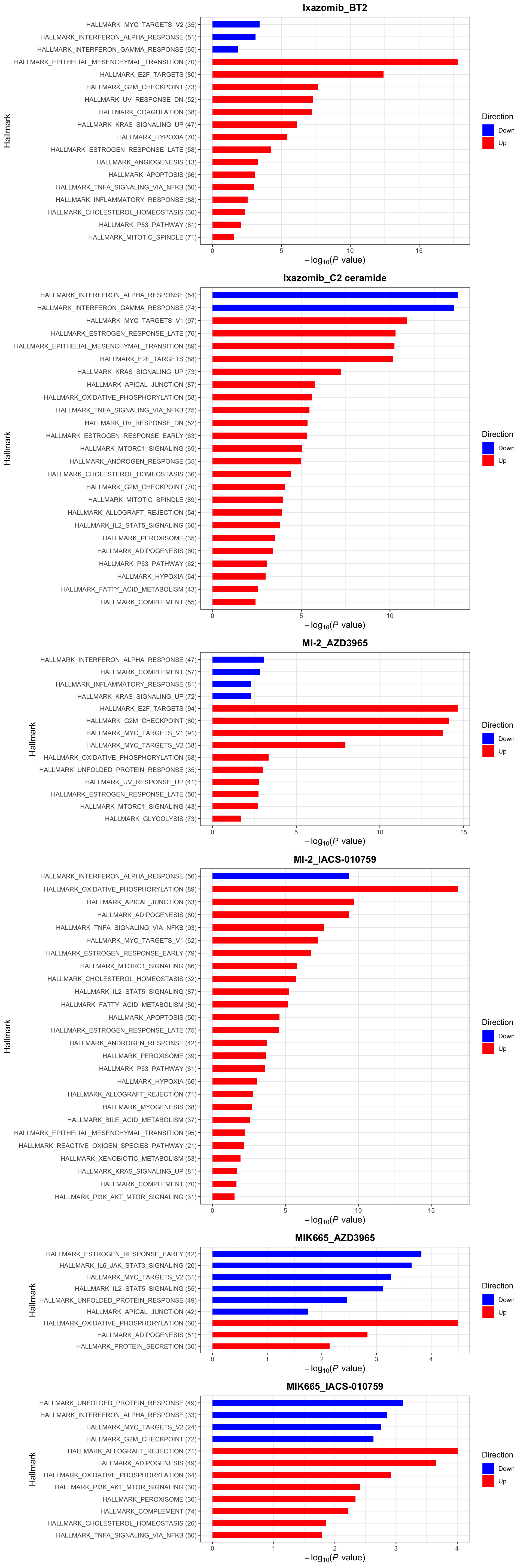
KEGG (10% FDR)
enrichres$KEGG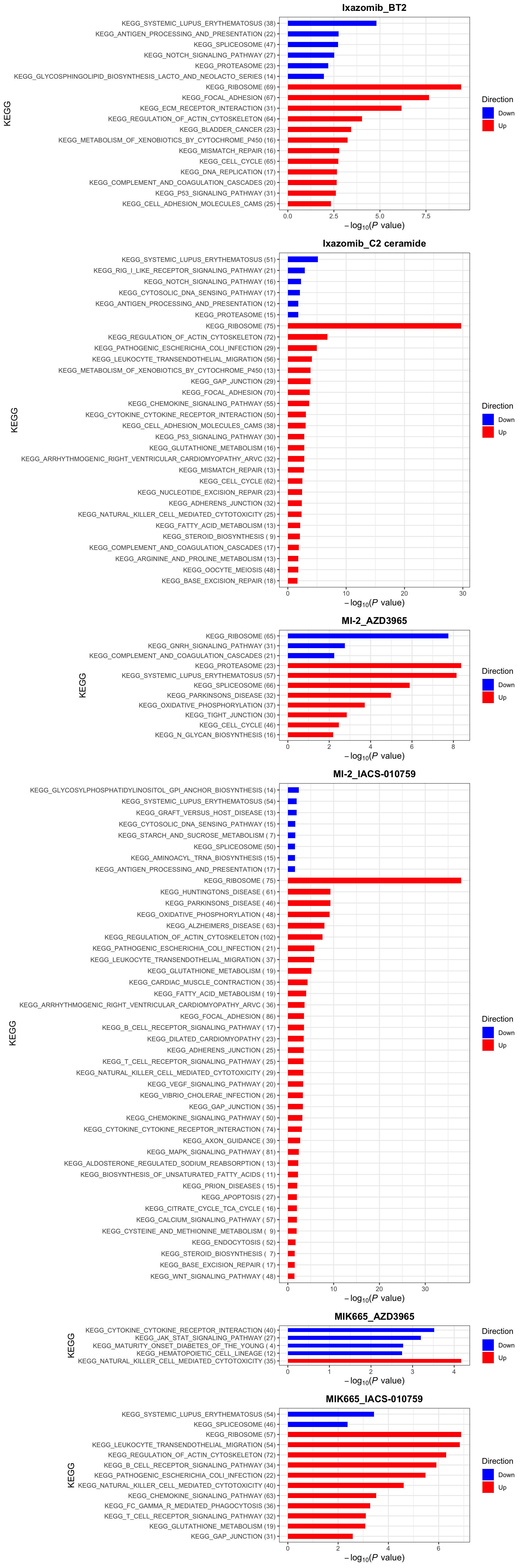
Explain synergy score by proteomics (from EMBL)
Processing proteomic data
Number of proteins and samples
load("../data/ProtWide.RData")
ProtWide <- ProtWide[,colnames(ProtWide) %in% unique(ciTabSum$Name)]
exprMat <- ProtWide
rownames(exprMat) <- getOneSymbol(rownames(exprMat), pos = "last", sep = "\\|")
#exprMat <- exprMat[!duplicated(rownames(exprMat)),]
dim(exprMat)[1] 4873 23Using Bliss synergy
Prepare list of selected synergy scores
synList <- ciTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination)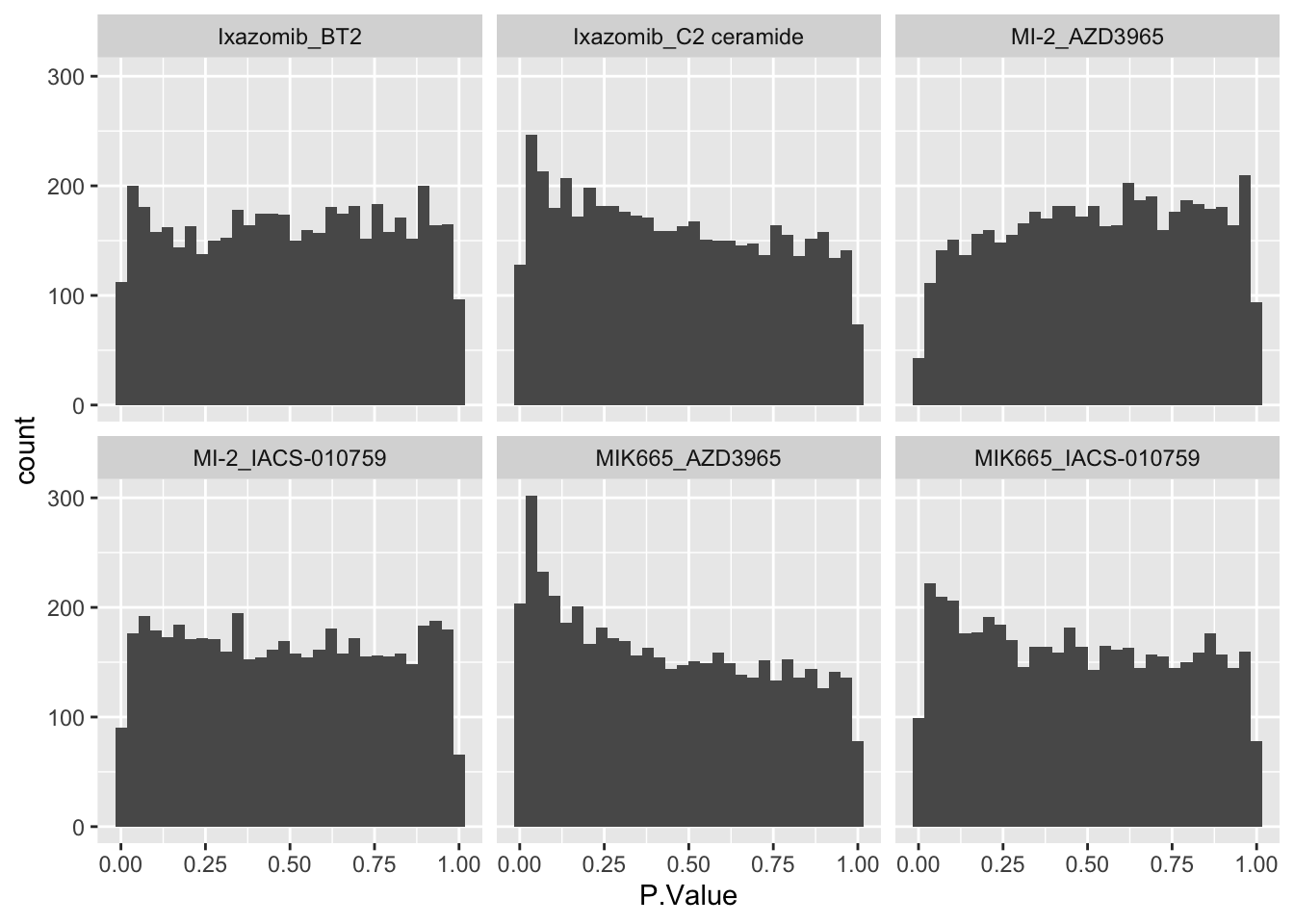 ### Number of significant associations
### Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 2
2 Ixazomib_C2 ceramide 0
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 0
5 MIK665_AZD3965 2
6 MIK665_IACS-010759 0Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 70
2 Ixazomib_C2 ceramide 61
3 MI-2_AZD3965 28
4 MI-2_IACS-010759 58
5 MIK665_AZD3965 129
6 MIK665_IACS-010759 56List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)[1] "No sets passed the criteria for MI-2_AZD3965 in KEGG"Cancer Hallmark (10% FDR)
enrichres$Hallmark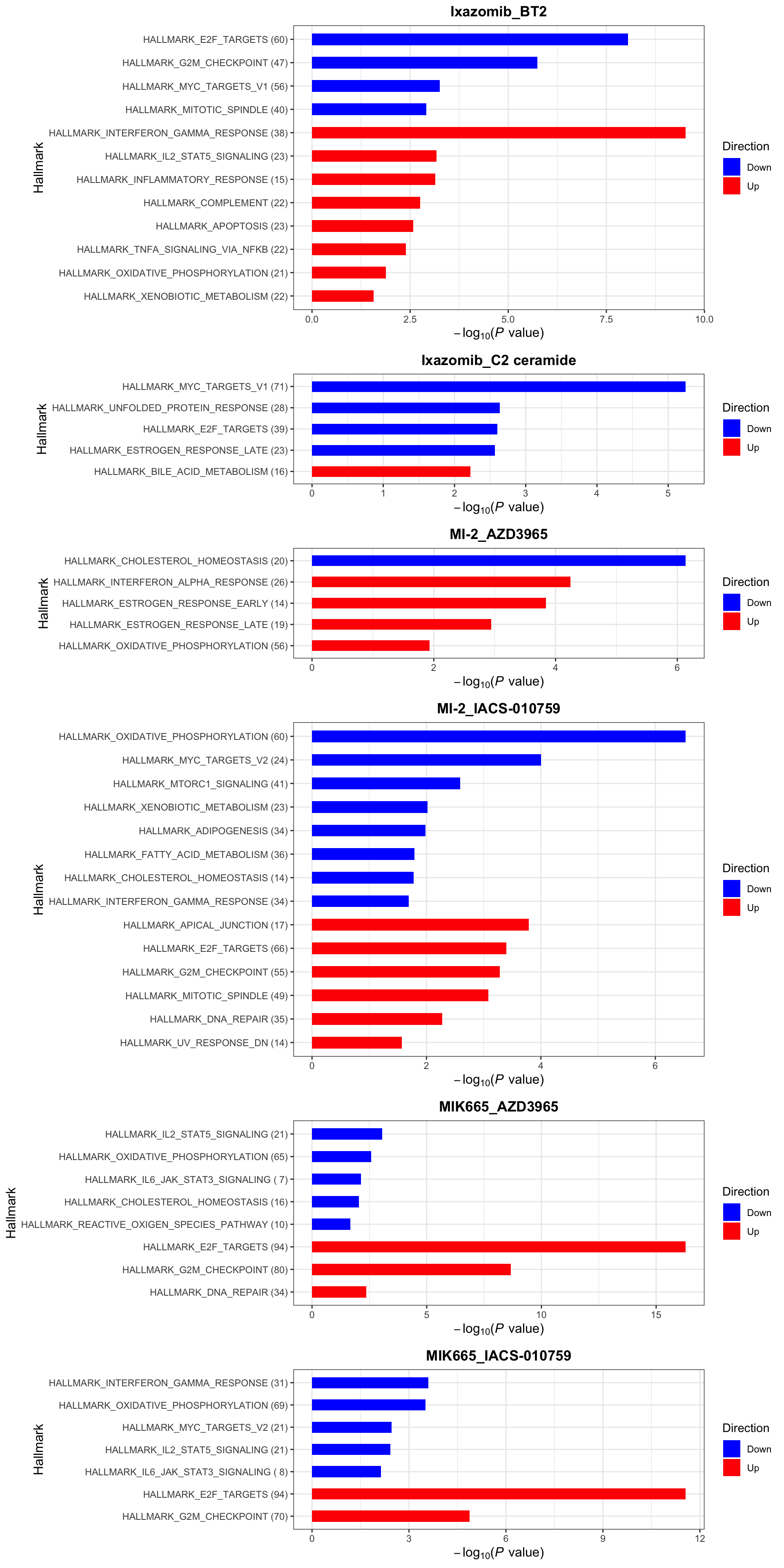
KEGG (10% FDR)
enrichres$KEGG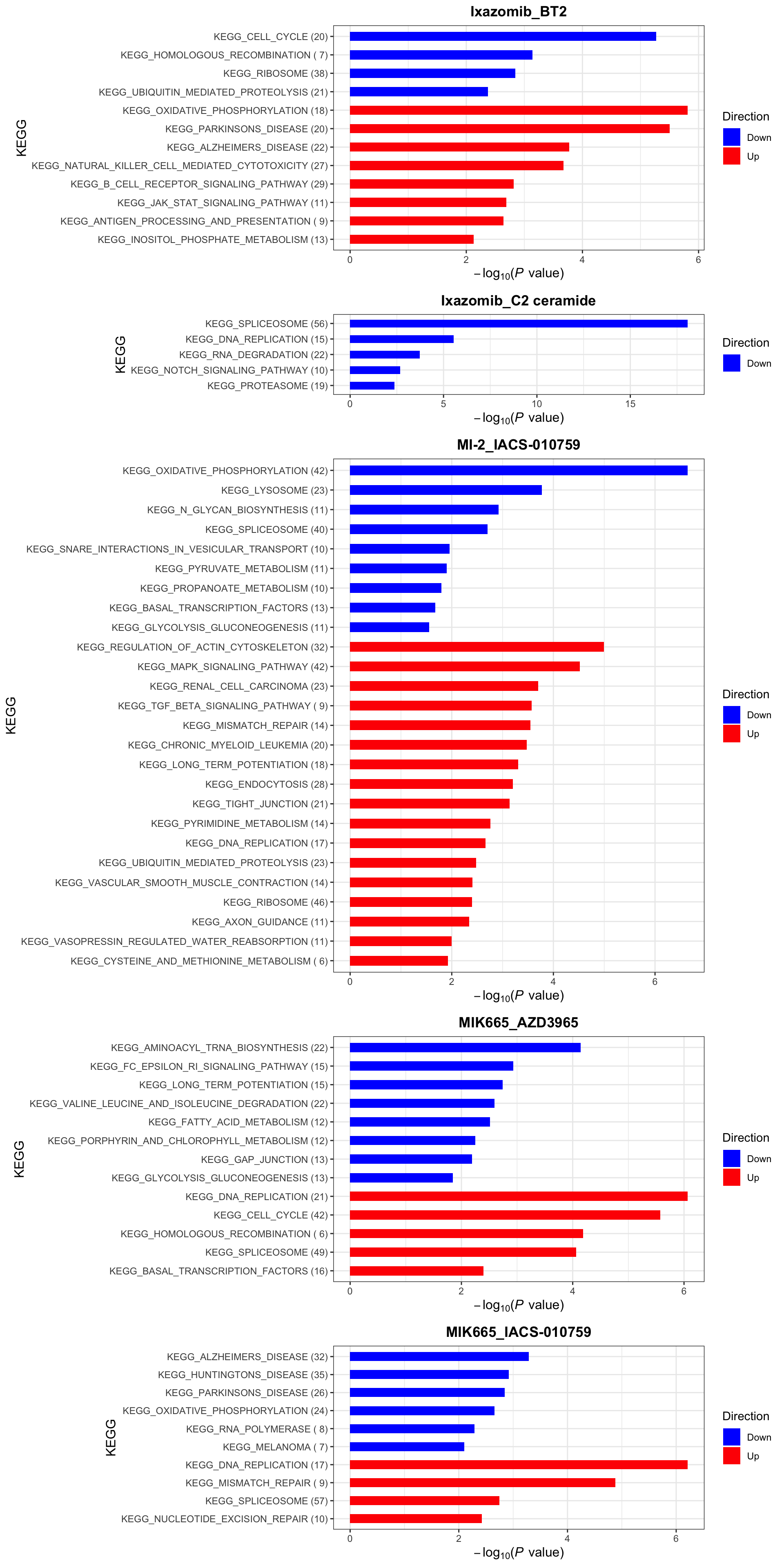
Using Bayes synergy
Prepare list of selected synergy scores
synList <- bayesTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination) ### Number of significant associations
### Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 2
2 Ixazomib_C2 ceramide 1
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 0
5 MIK665_AZD3965 4
6 MIK665_IACS-010759 1Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 53
2 Ixazomib_C2 ceramide 45
3 MI-2_AZD3965 121
4 MI-2_IACS-010759 40
5 MIK665_AZD3965 180
6 MIK665_IACS-010759 98List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)Cancer Hallmark (10% FDR)
enrichres$Hallmark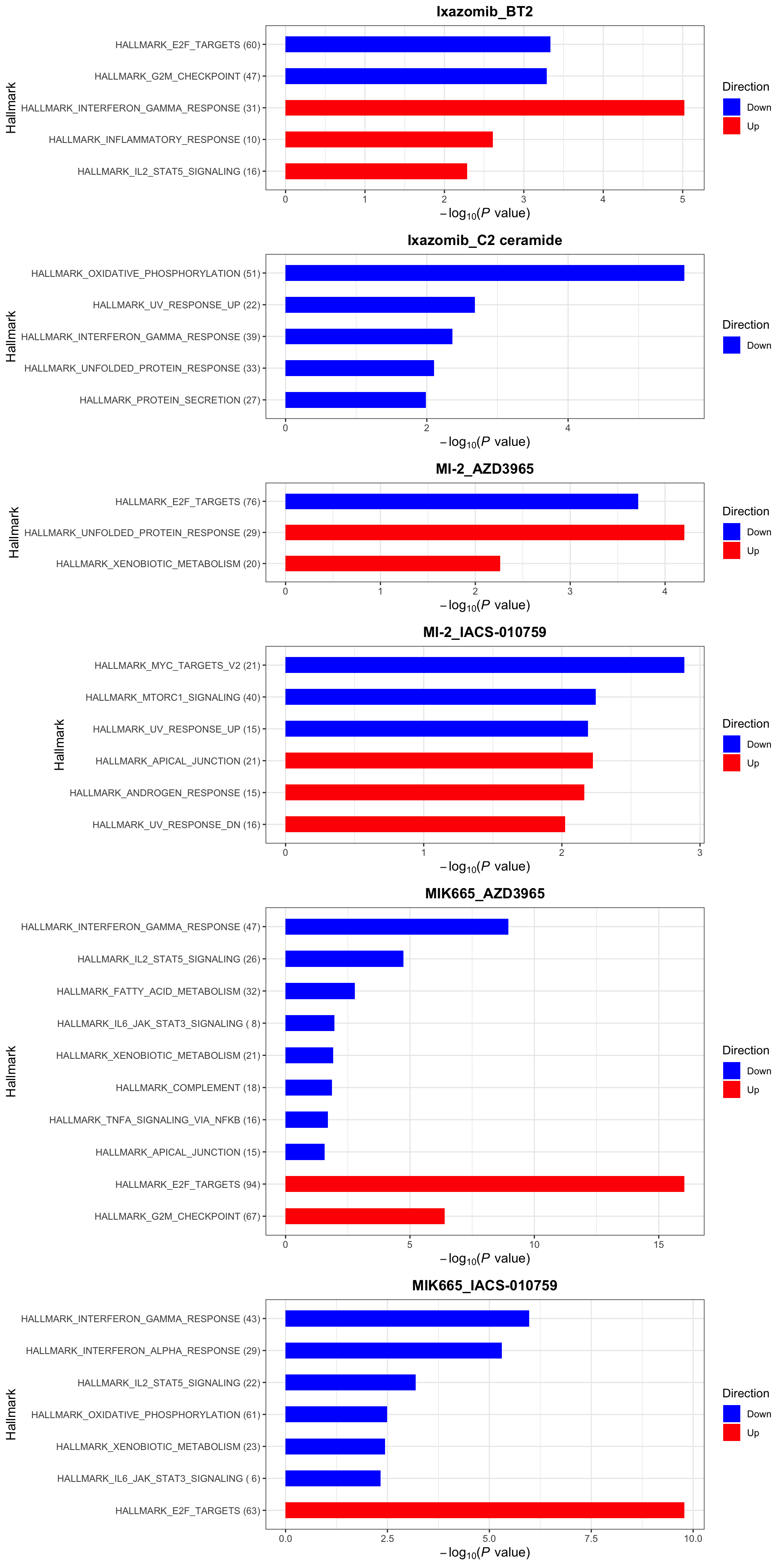
KEGG (10% FDR)
enrichres$KEGG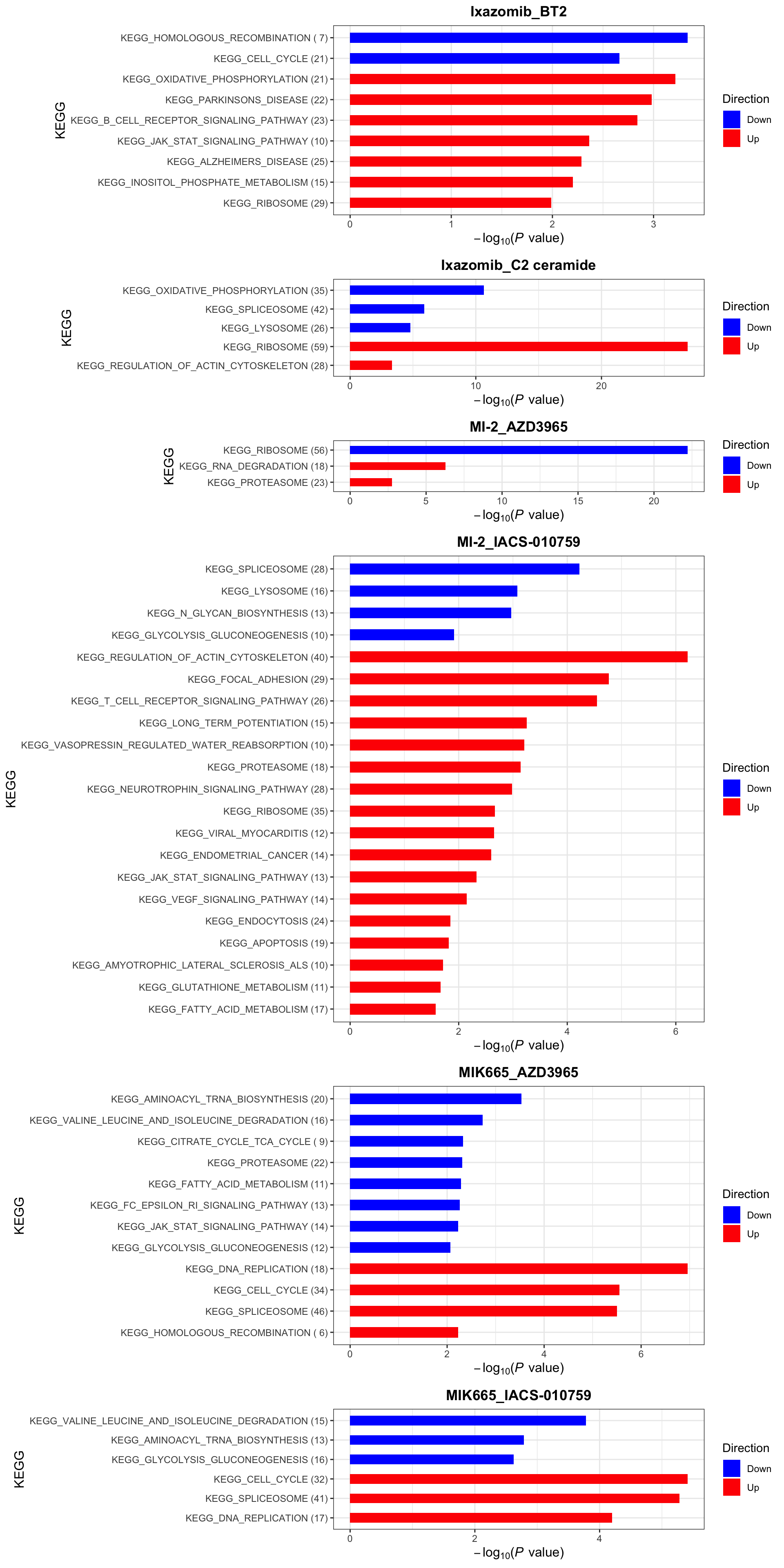
Explain synergy score by proteomics (from AG Krijgsveld)
Processing proteomic data
library(SummarizedExperiment)
protData <- readRDS("../data/SC005_SummarizedExperiment_proteomics.RDS")
#select baseline samples
protData <- protData[!rowData(protData)$Gene_name %in% c("",NA), protData$cell.line %in% unique(ciTabSum$Name) & protData$condition == "U"]
rowData(protData)$Gene_name <- getOneSymbol(rowData(protData)$Gene_name, pos = "first")
assayNames(protData) <- "norm"
protTab <- jyluMisc::sumToTidy(protData) %>%
group_by(cell.line, Gene_name) %>%
summarise(count = mean(norm, na.rm=TRUE)) %>%
dplyr::rename(symbol = Gene_name, cellLine = cell.line) %>%
mutate(colID = cellLine) %>%
ungroup()
protData <- jyluMisc::tidyToSum(protTab, rowID = "symbol", colID = "colID",
values = "count", annoRow = "symbol",
annoCol = c("cellLine"))
exprMat <- assay(protData)
#rownames(exprMat) <- rowData(protData)$symbol
dim(exprMat)[1] 2640 11Using Bliss synergy
Prepare list of selected synergy scores
synList <- ciTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination) ### Number of significant associations
### Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 15
2 Ixazomib_C2 ceramide 0
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 1
5 MIK665_AZD3965 1
6 MIK665_IACS-010759 0Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 60
2 Ixazomib_C2 ceramide 11
3 MI-2_AZD3965 16
4 MI-2_IACS-010759 47
5 MIK665_AZD3965 74
6 MIK665_IACS-010759 37List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)[1] "No sets passed the criteria for Ixazomib_C2 ceramide in Hallmark"Cancer Hallmark (10% FDR)
enrichres$Hallmark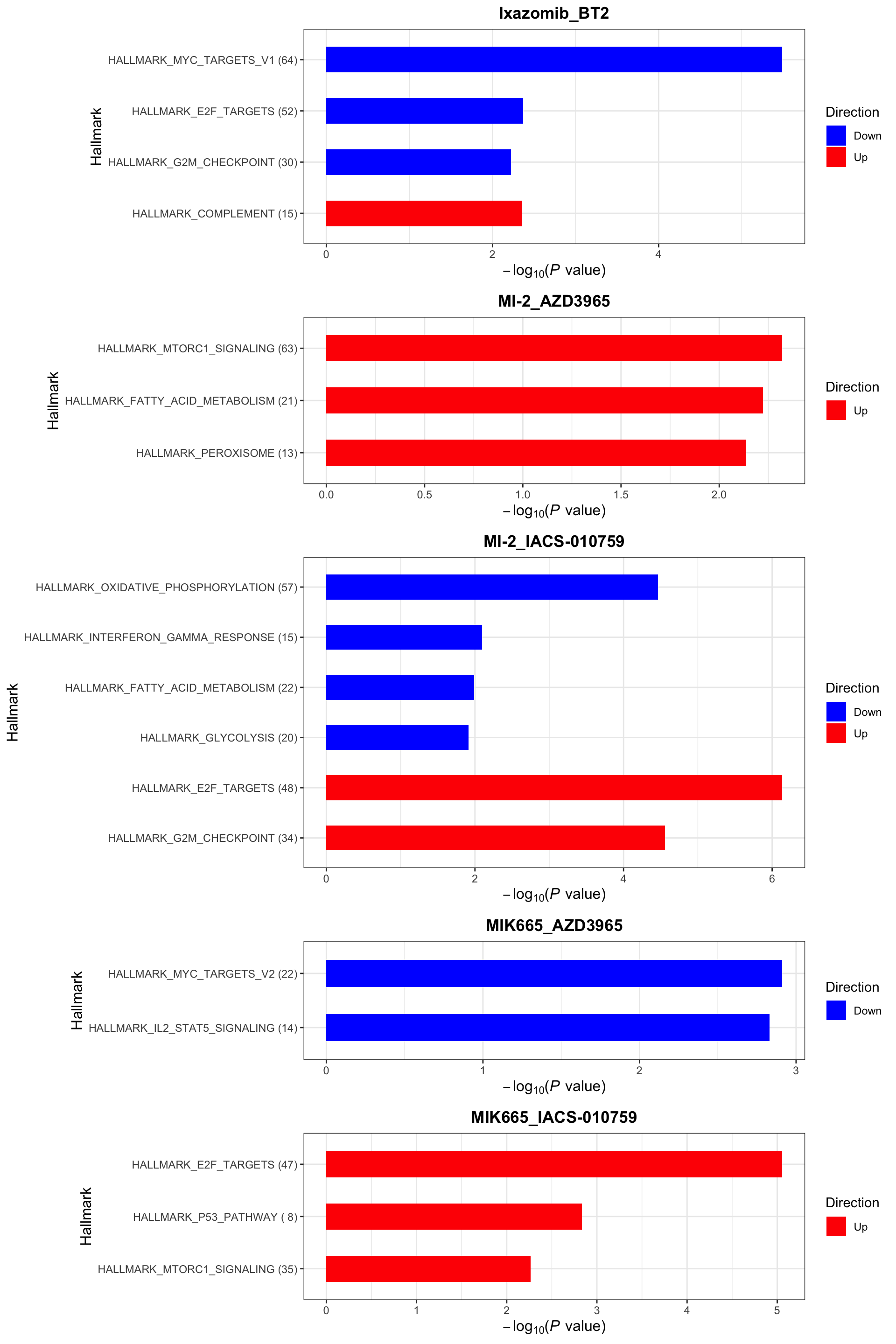
KEGG (10% FDR)
enrichres$KEGG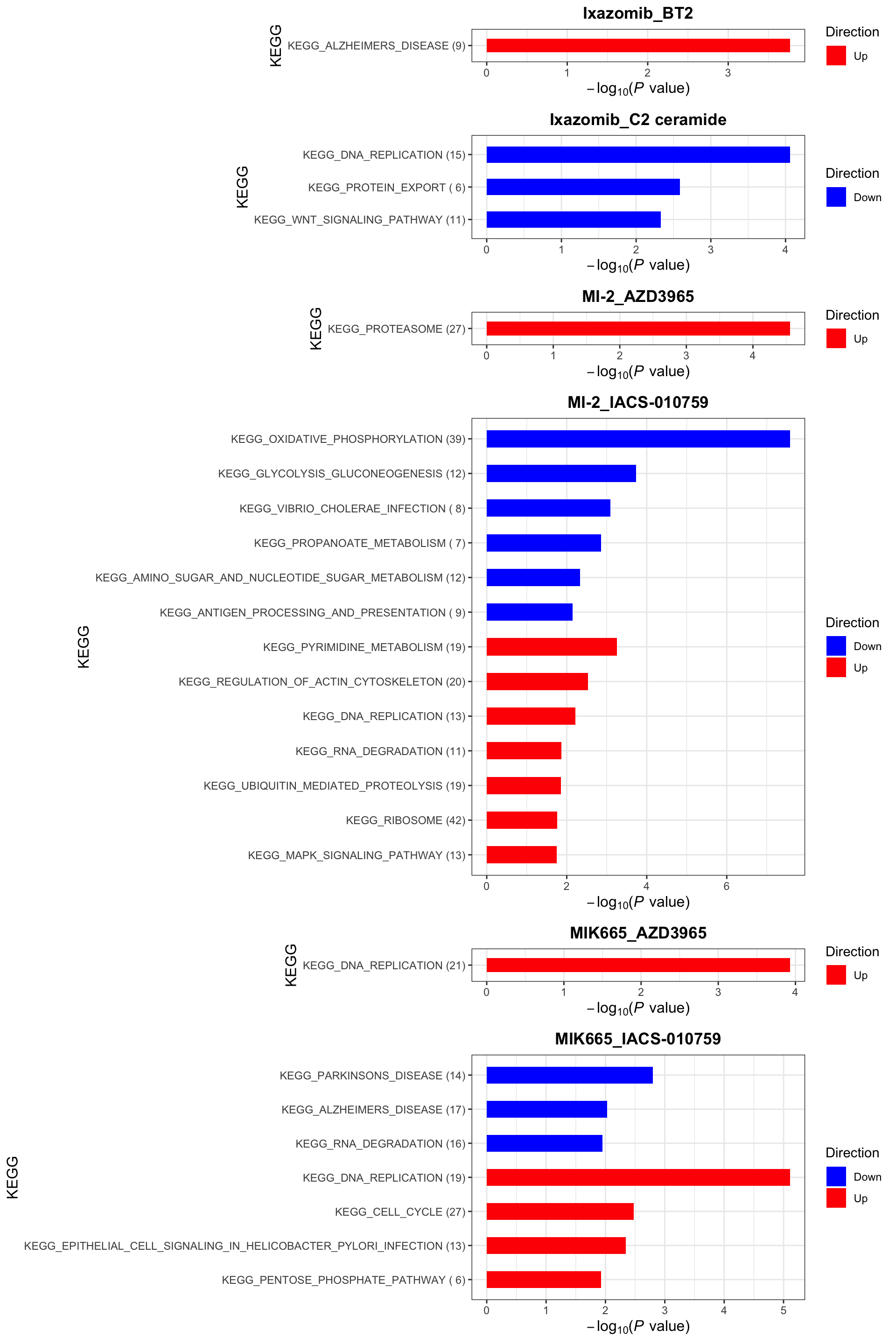
Using Bayes synergy
Prepare list of selected synergy scores
synList <- bayesTabSum %>%
mutate(combination = paste0(Drug_A, "_", Drug_B)) %>%
filter(combination %in% paste0(selectedPair$Drug_A,"_",selectedPair$Drug_B)) %>%
mutate(score = syn)Perform association tests
allRes <- predictSynergy(synList, exprMat)P-value histogram
ggplot(allRes, aes(x=P.Value)) +
geom_histogram() +
facet_wrap(~combination)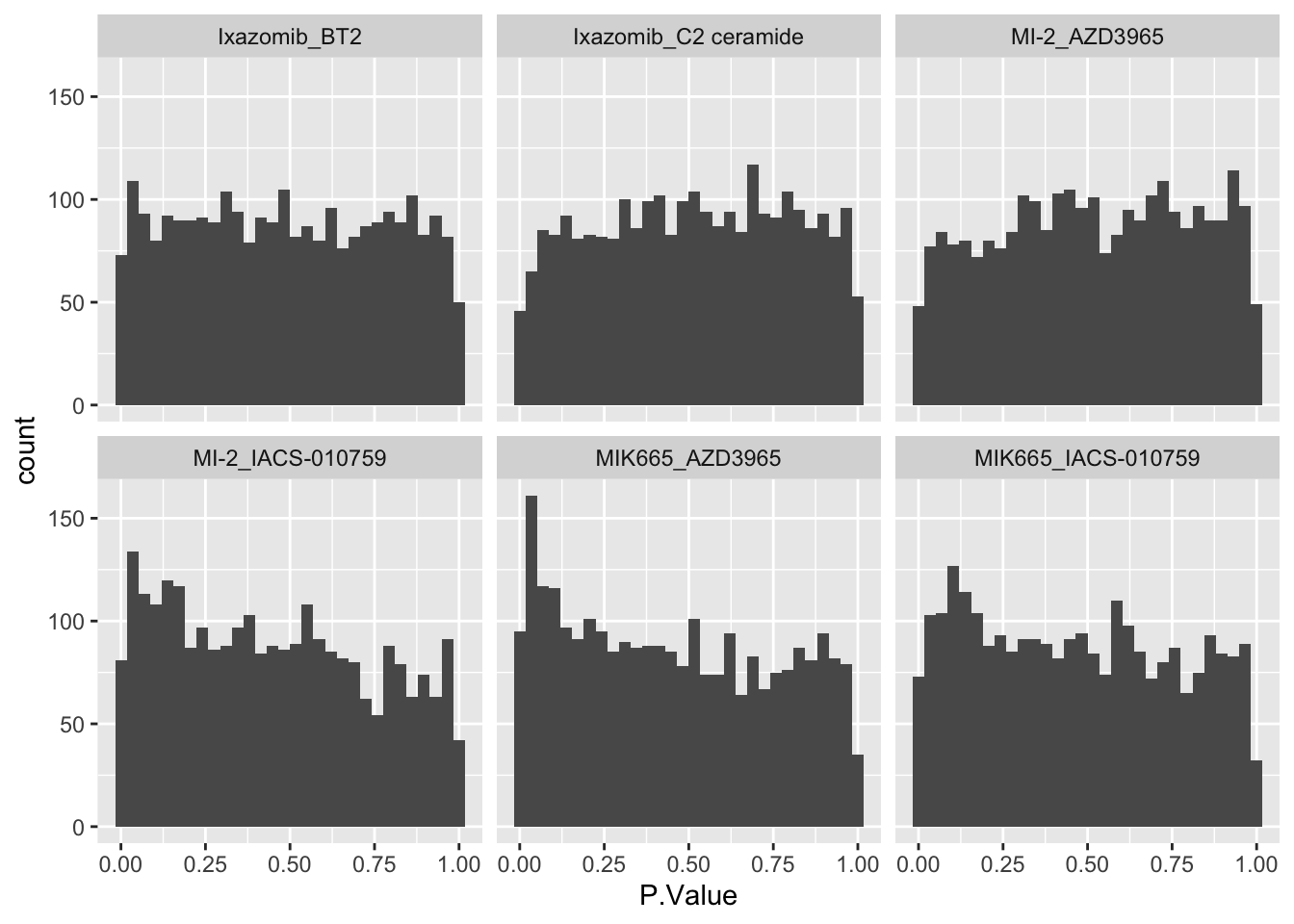 ### Number of significant associations
### Number of significant associations
Use 10% FDR (adj.P.Val < 0.1)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(adj.P.Val <= 0.10))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 3
2 Ixazomib_C2 ceramide 0
3 MI-2_AZD3965 0
4 MI-2_IACS-010759 0
5 MIK665_AZD3965 2
6 MIK665_IACS-010759 1Use raw P.Value < 0.01 (without multiple hypothesis adjustment)
sumTab <- group_by(allRes, combination) %>%
summarise(num = sum(P.Value <= 0.01))
sumTab# A tibble: 6 × 2
combination num
<chr> <int>
1 Ixazomib_BT2 55
2 Ixazomib_C2 ceramide 25
3 MI-2_AZD3965 28
4 MI-2_IACS-010759 50
5 MIK665_AZD3965 60
6 MIK665_IACS-010759 50List of significantly associated genes (raw P < 0.01)
allRes.sig <- filter(allRes, P.Value < 0.01) %>%
select(combination, symbol, logFC, P.Value, adj.P.Val) %>%
mutate_if(is.numeric, formatC, digits=1)
DT::datatable(allRes.sig)Pathway Enrichment analysis
gmts = list(Hallmark= "../data/gmts/h.all.v6.2.symbols.gmt",
KEGG = "../data/gmts/c2.cp.kegg.v6.2.symbols.gmt")
enrichres <- performEnrichmentOnList(allRes, gmts, pCut = 0.1, ifFDR = TRUE)[1] "No sets passed the criteria for MI-2_AZD3965 in Hallmark"
[1] "No sets passed the criteria for MI-2_IACS-010759 in Hallmark"
[1] "No sets passed the criteria for Ixazomib_BT2 in KEGG"Cancer Hallmark (10% FDR)
enrichres$Hallmark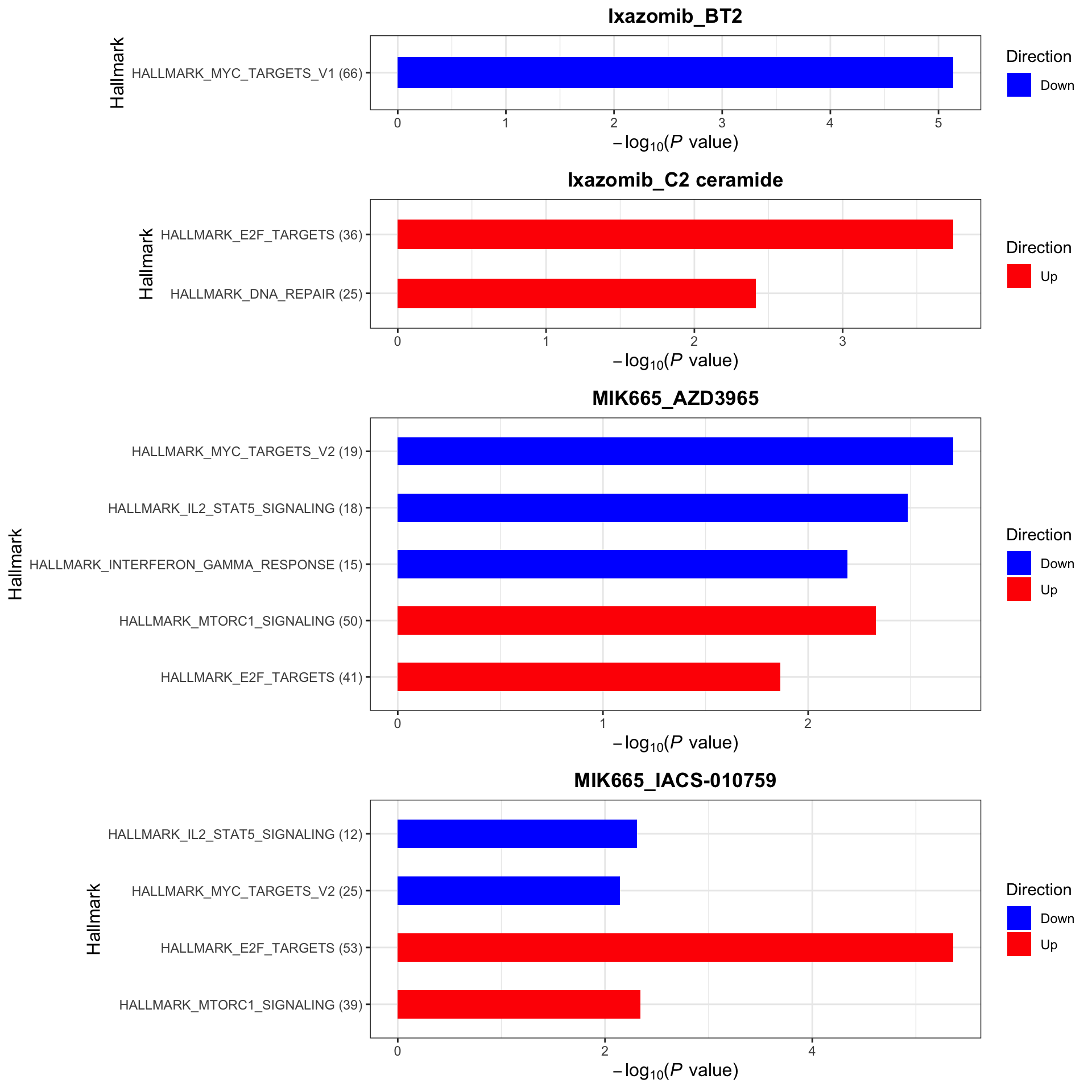
KEGG (10% FDR)
enrichres$KEGG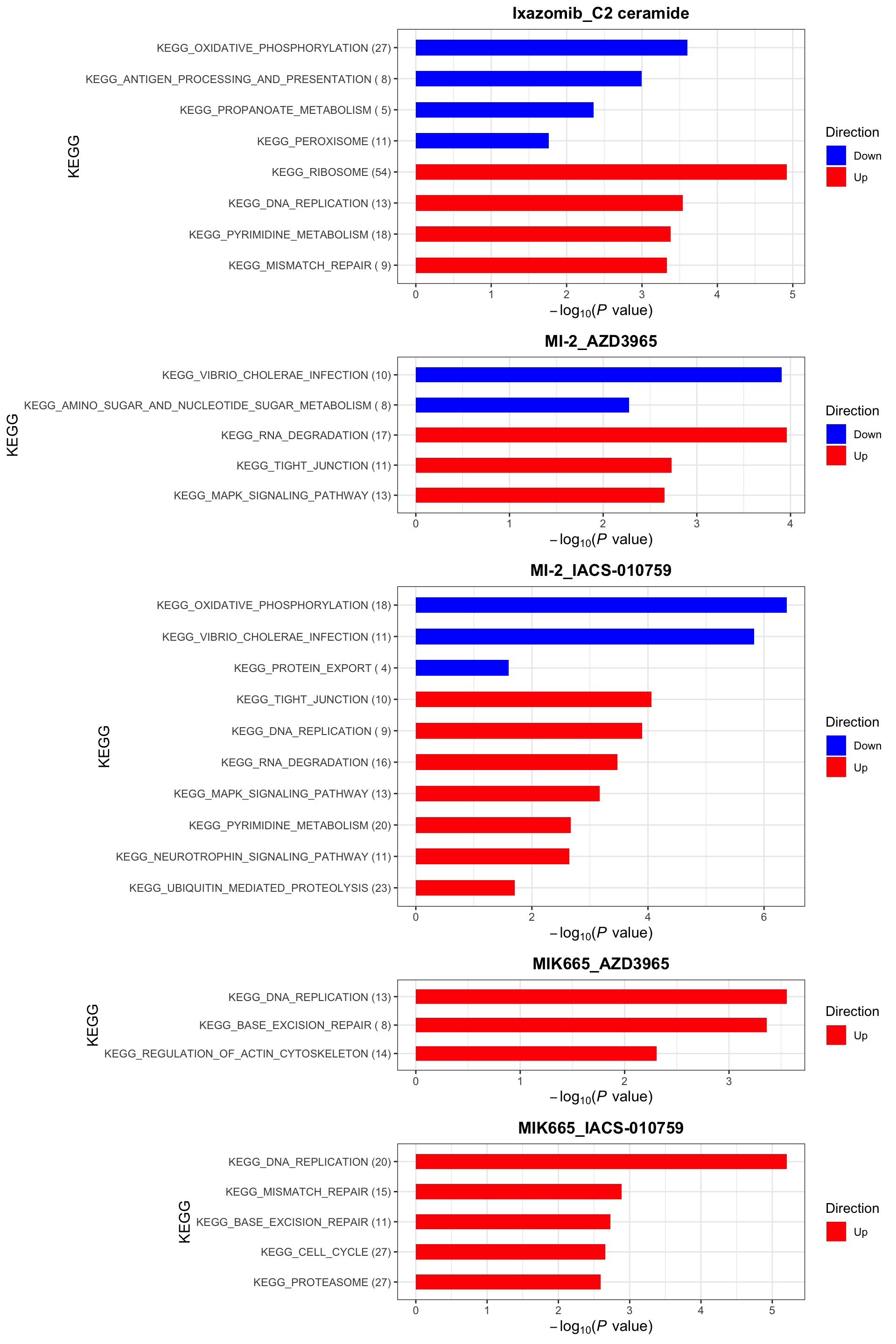
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur/Monterey 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] SummarizedExperiment_1.26.1 Biobase_2.56.0
[3] GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[5] IRanges_2.30.0 S4Vectors_0.34.0
[7] BiocGenerics_0.42.0 MatrixGenerics_1.8.1
[9] matrixStats_0.62.0 gridExtra_2.3
[11] forcats_0.5.1 stringr_1.4.1
[13] dplyr_1.0.9 purrr_0.3.4
[15] readr_2.1.2 tidyr_1.2.0
[17] tibble_3.1.8 ggplot2_3.4.1
[19] tidyverse_1.3.2
loaded via a namespace (and not attached):
[1] readxl_1.4.0 backports_1.4.1 fastmatch_1.1-3
[4] drc_3.0-1 jyluMisc_0.1.5 workflowr_1.7.0
[7] igraph_1.3.4 shinydashboard_0.7.2 splines_4.2.0
[10] crosstalk_1.2.0 BiocParallel_1.30.3 TH.data_1.1-1
[13] digest_0.6.30 htmltools_0.5.4 fansi_1.0.3
[16] magrittr_2.0.3 googlesheets4_1.0.0 cluster_2.1.3
[19] tzdb_0.3.0 limma_3.52.2 modelr_0.1.8
[22] sandwich_3.0-2 piano_2.12.0 colorspace_2.0-3
[25] ggrepel_0.9.1 rvest_1.0.2 haven_2.5.0
[28] xfun_0.31 crayon_1.5.2 RCurl_1.98-1.7
[31] jsonlite_1.8.3 survival_3.4-0 zoo_1.8-10
[34] glue_1.6.2 survminer_0.4.9 gtable_0.3.0
[37] gargle_1.2.0 zlibbioc_1.42.0 XVector_0.36.0
[40] DelayedArray_0.22.0 car_3.1-0 abind_1.4-5
[43] scales_1.2.0 vsn_3.64.0 mvtnorm_1.1-3
[46] DBI_1.1.3 relations_0.6-12 rstatix_0.7.0
[49] Rcpp_1.0.9 plotrix_3.8-2 xtable_1.8-4
[52] preprocessCore_1.58.0 km.ci_0.5-6 DT_0.23
[55] htmlwidgets_1.5.4 httr_1.4.3 fgsea_1.22.0
[58] gplots_3.1.3 ellipsis_0.3.2 pkgconfig_2.0.3
[61] farver_2.1.1 sass_0.4.2 dbplyr_2.2.1
[64] utf8_1.2.2 labeling_0.4.2 tidyselect_1.1.2
[67] rlang_1.0.6 later_1.3.0 munsell_0.5.0
[70] cellranger_1.1.0 tools_4.2.0 visNetwork_2.1.0
[73] cachem_1.0.6 cli_3.4.1 generics_0.1.3
[76] broom_1.0.0 evaluate_0.15 fastmap_1.1.0
[79] yaml_2.3.5 knitr_1.39 fs_1.5.2
[82] survMisc_0.5.6 caTools_1.18.2 nlme_3.1-158
[85] mime_0.12 slam_0.1-50 xml2_1.3.3
[88] compiler_4.2.0 rstudioapi_0.13 affyio_1.66.0
[91] ggsignif_0.6.3 marray_1.74.0 reprex_2.0.1
[94] bslib_0.4.1 stringi_1.7.8 highr_0.9
[97] lattice_0.20-45 Matrix_1.5-4 KMsurv_0.1-5
[100] shinyjs_2.1.0 vctrs_0.5.2 pillar_1.8.0
[103] lifecycle_1.0.3 BiocManager_1.30.18 jquerylib_0.1.4
[106] data.table_1.14.8 cowplot_1.1.1 bitops_1.0-7
[109] httpuv_1.6.6 affy_1.74.0 R6_2.5.1
[112] promises_1.2.0.1 KernSmooth_2.23-20 codetools_0.2-18
[115] MASS_7.3-58 gtools_3.9.3 exactRankTests_0.8-35
[118] assertthat_0.2.1 rprojroot_2.0.3 withr_2.5.0
[121] multcomp_1.4-19 GenomeInfoDbData_1.2.8 mgcv_1.8-40
[124] parallel_4.2.0 hms_1.1.1 grid_4.2.0
[127] rmarkdown_2.14 carData_3.0-5 googledrive_2.0.0
[130] git2r_0.30.1 maxstat_0.7-25 ggpubr_0.4.0
[133] sets_1.0-21 shiny_1.7.4 lubridate_1.8.0