Approach & Outputs
The overarching concept is constructing low-dimensional representations of high-dimensional biological data to understand complex biological systems using interpretable mathematical modeling.
- Representation learning for biology in space and time: bring together different data types and resolutions to find low-dimensional explanations (factors, gradients, regions, graphs and networks) of high-dimensional data, using statistical models, first-principles based theory and machine learning.
- Spatial omics in immunooncology and systems immunology: find and improve treatment options for patients.
- Multidimensional phenotypes: map context-dependent gene-gene and gene-drug interaction networks from genetic or drug-based perturbation assays.
- Translational statistics: many powerful mathematical and computational ideas exist but are difficult to access. We aim to translate them into practical methods and software that make a real difference to biomedical researchers.

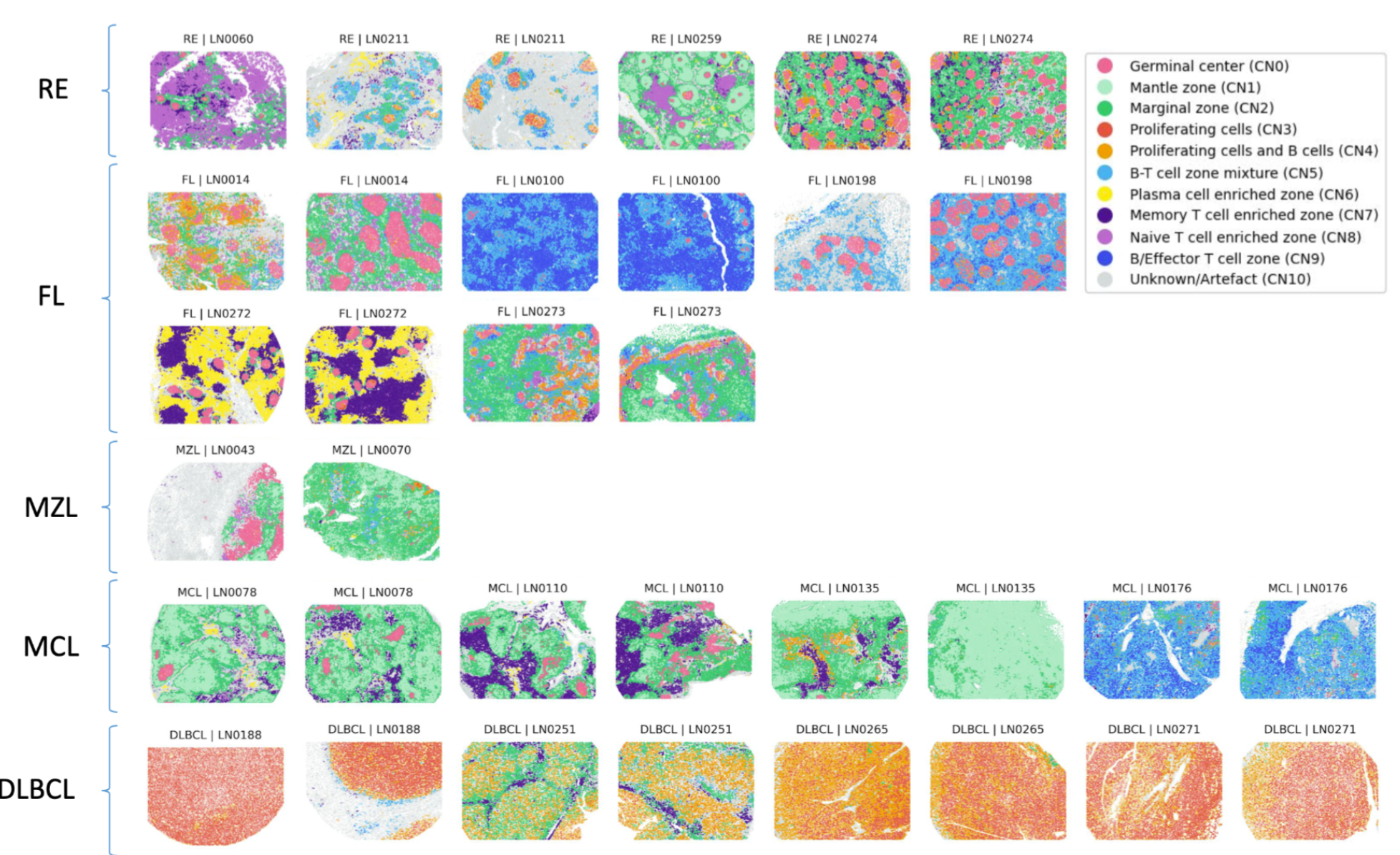
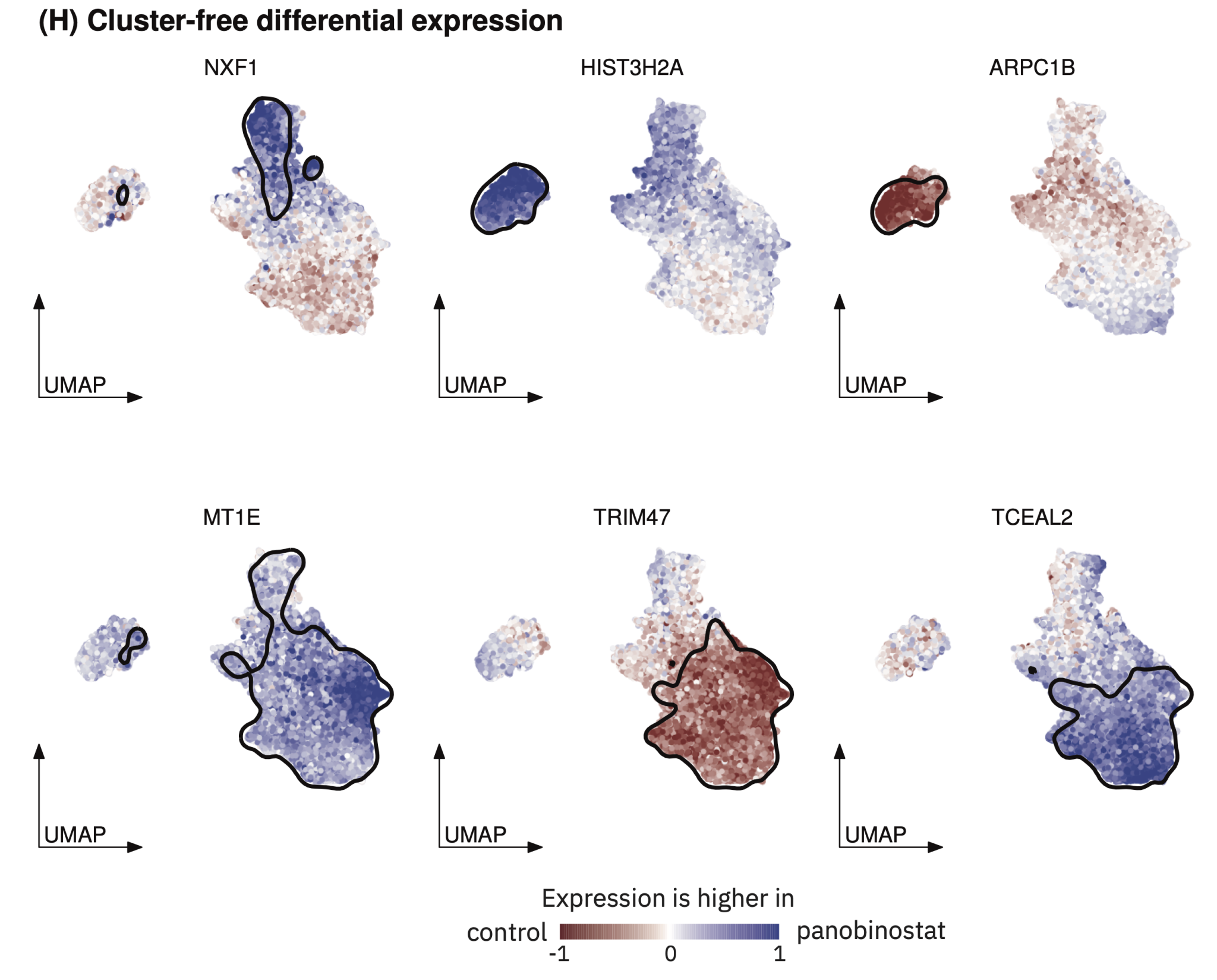

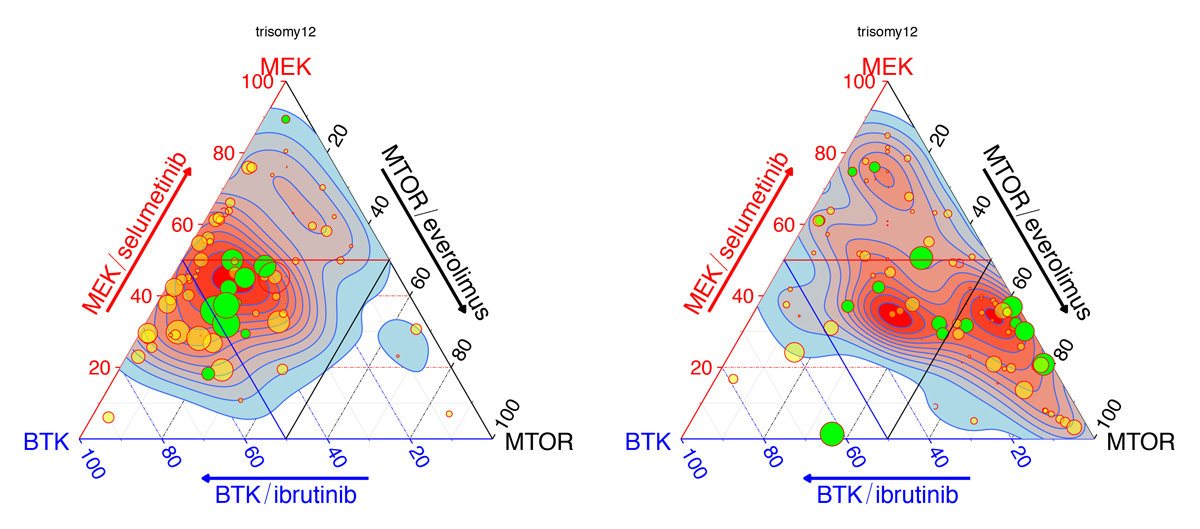
Spatial omics and imaging
Spatial omics techniques measure the spatial distribution of thousands distinct molecules, but are limited by sampling efficiency and spatial resolution. Photon and electron microscopy can reach resolutions of tens of nanometers or even Ångströms, but only for one or a few distinct fluorophores, or physical properties of matter. How to bring this all together? How to navigate huge Terabyte-scale maps of cells and organisms? Then, beyond static maps, how to decipher differential activities of biological functions and processes, associate them with phenotypes in health and disease, and exploit this for better understanding fundamental biology and advance biotechnology and biomedicine?
Functional precision medicine and immuno-oncology
We integrate observational ’omics data, interventional clinical data, and systematic genetic or chemical perturbation data on (ex-vivo) model systems to decipher the molecular mechanisms of variable sensitivity and resistance of tumors to treatments (precision oncology collaboration with Thorsten Zenz at University Hospital Zurich), and to understand the role of the immune system and the tumour microenvironment in tumourigenesis, progression and treatment (systems immunology collaboration with Sascha Dietrich at University Hospital Düsseldorf).
Method development and discovery science
We pursue these hand-in-hand: we engage in biological and biomedical discovery science projects (typically through collaborations) to study new datasets and questions, use these to derive new challenges for analytical method development and conceptual modelling, and then test, evaluate and harden our theoretical and computational approaches on these real-life challenges.
Open science
As we engage with new data types, we aim to develop high-quality computational methods of wide applicability. We consider the release and maintenance of scientific software an integral part of doing science. We contribute to the Bioconductor project, an open source software collaboration to provide tools for the analysis and understanding of genome-scale data. An example is our DESeq2 package for analyzing count data from high-throughput sequencing.
Mentoring and career development
Science is an intellectual adventure and a creative process done by people. For each of us, our work is at the same time, a means to achieve a scientific goal, a job that enables us to pay our bills, and a stage of training and professional development. This includes internships, BSc/MSc theses, PhD theses, postdoctoral projects. The group, and EMBL more generally, offers a well-established mentoring framework to support these triple objectives. Former group members have moved on to rewarding careers: professors, independent group leaders, senior management or professional scientist roles in industry.
Teaching
We maintain the textbook Modern Statistics for Modern Biology by Susan Holmes and Wolfgang Huber. The book is available online, for free, as HTML. It was published as a printed book in 2019 by Cambridge University Press.
We run the annual summer school CSAMA—Biological Data Science. It usually takes place in June in Brixen/Bressanone. Here is the webpage of the 2026 edition. See here for some impressions.
In July 2023, 2024, 2025 and 2026, we co-organized the Ukrainian Biological Data Science Summer School in Uzhhorod, Ukraine. See also Wolfgang’s post about it.
We develop publicly available interactive training materials on statistical methods.
Software
We are a frequent contributor to the Bioconductor project
| LEMUR | Cluster-free differential expression analysis of multi-condition single-cell data using Latent Embedding Multivariate Regression |
| MOFA | Multi-Omics Factor Analysis |
| DESeq2 | Differential gene expression analysis based on the negative binomial distribution |
| IHW | Multiple testing and false discovery rate (FDR) control by Independent Hypothesis Weighting |
| EBImage | Image processing and analysis toolbox for R |
| Rarr | Read Zarr Files in R |
| rhdf5 | R Interface to HDF5 |
| vsn | Normalization and variance stabilizing transformation of fluorescence intensity data |
| cellHTS2 | Analysis of cell-based high-throughput screens |
| DEXSeq | Inference of differential exon usage in RNA-Seq |
| HilbertVis | Visualize long vectors of data using Hilbert curves |
| Python | |
| HTSeq | Processing and analyzing data from high-throughput sequencing assays |
| SOFA | Semi-supervised (Multi) Omics Factor Analysis |
| spatialproteomics | lightweight wrapper around xarray to facilitate processing, exploration and analysis of multiplexed immunohistochemistry data |